Las plaquetas son pequeñas células en forma de disco que circulan en la sangre. Desempeñan un papel importante en la formación de coágulos sanguíneos para ayudar a detener hemorragias y en la reparación de vasos sanguíneos dañados.
Cuando un vaso sanguíneo se lesiona, las plaquetas empiezan el proceso para detener la hemorragia formando lo que se conoce como un tapón plaquetario. Esto tiene lugar en tres etapas (véase la figura 1):
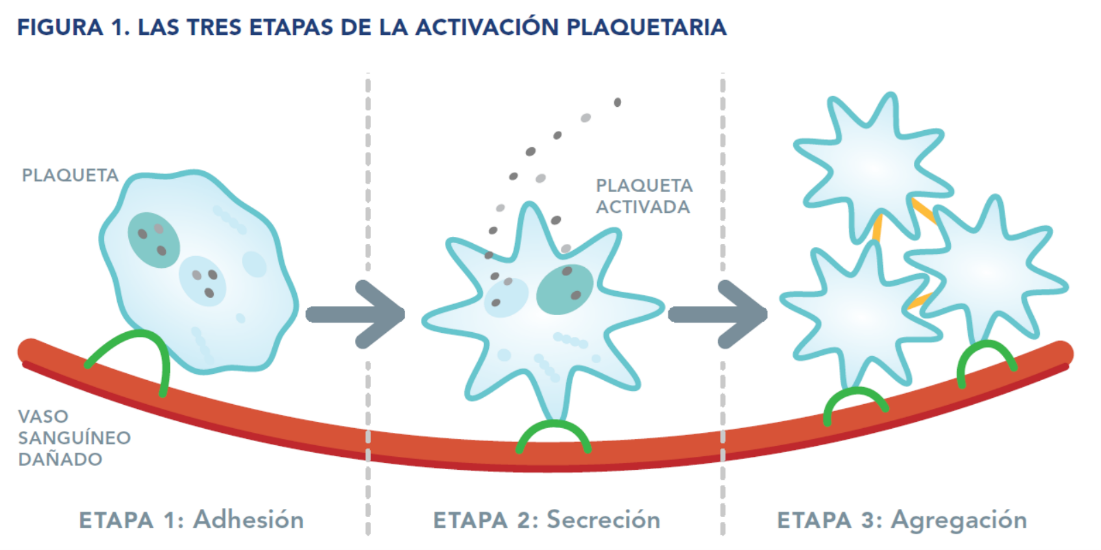
Conforme se forma el tapón plaquetario, también se reclutan al lugar de la lesión unas proteínas llamadas factores de coagulación. Estos factores de coagulación trabajan en conjunto sobre la superficie de las plaquetas para fortalecer el tapón plaquetario formando una red llamada coágulo de fibrina.
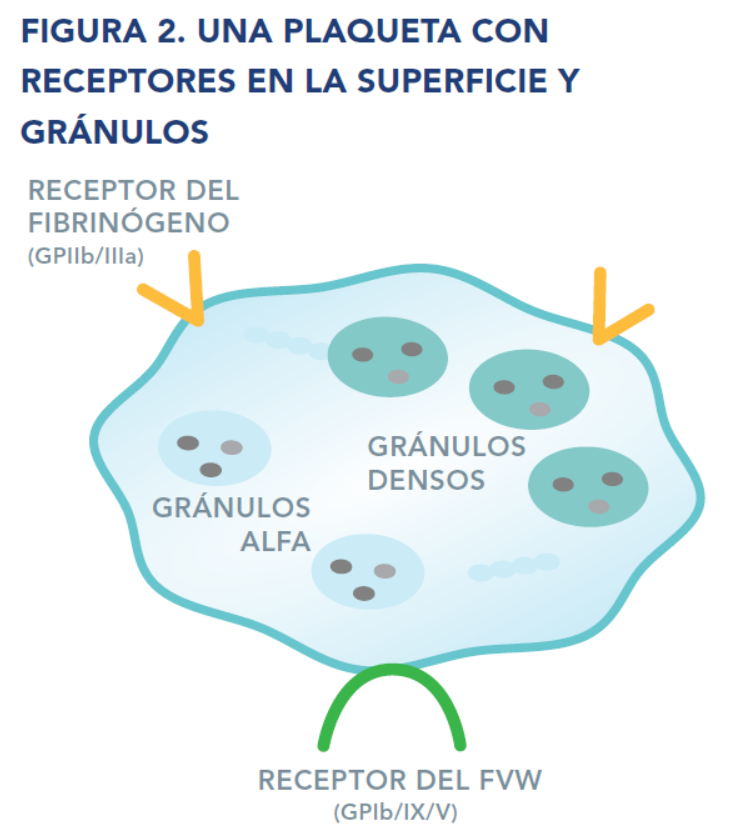
Las plaquetas tienen varios componentes, tales como receptores en la superficie y gránulos internos, los cuales contribuyen a la formación del coágulo.
Receptores
Los receptores son proteínas en la superficie de las plaquetas que ayudan a éstas a interactuar con sustancias químicas, y otras proteínas o células de la sangre, y a responder ante ellas.
Gránulos
Los gránulos son pequeños paquetes al interior de las plaquetas en los que se almacenan proteínas y otras sustancias químicas. El contenido de los gránulos se libera de las plaquetas durante la fase de secreción de la activación. Estas moléculas funcionan como señales para reclutar más plaquetas y otras células al lugar de la lesión a fin de detener la hemorragia.
Hay dos tipos de gránulos: los gránulos alfa y los gránulos densos. Los contenidos de los gránulos alfa y de los gránulos densos son diferentes y funcionan de diferentes maneras para reclutar más plaquetas, activar a los factores de la coagulación, y detener la hemorragia.
Los trastornos de la función plaquetaria son padecimientos en los que las plaquetas no funcionan como deberían. Si el tapón plaquetario no se forma adecuadamente, la hemorragia puede continuar durante más tiempo de lo normal. Las personas que padecen trastornos de la función plaquetaria tienden a presentar moretones y hemorragias más fácilmente de lo normal. Los trastornos de la función plaquetaria pueden ser provocados por un problema con uno de los receptores, con los gránulos, o con los procesos de activación al interior de las plaquetas, y pudieran no estar relacionados con un conteo plaquetario bajo.
Hay trastornos de la función plaquetaria hereditarios (es decir, que se transmiten de padres a hijos) y adquiridos. Esta monografía aborda los trastornos hereditarios de la función plaquetaria.
El síndrome de Bernard-Soulier es un trastorno hereditario de la función plaquetaria causado por una anomalía en el receptor del factor Von Willebrand (FVW, véase la figura 2). Este receptor también es llamado receptor de la glicoproteína (GP) Ib/IX/V. Los receptores son proteínas en la superficie de las plaquetas que las ayudan a interactuar con otras células o sustancias sanguíneas y a responder ante ellas. Dado que el receptor del FVW está ausente o no funciona adecuadamente, las plaquetas no pueden unirse al FVW y no se adhieren a la pared del vaso sanguíneo lesionado del modo en que deberían hacerlo. El resultado es que el tapón plaquetario no se forma normalmente.
El síndrome de Bernard-Soulier es un trastorno autosómico recesivo, lo que quiere decir que ambos padres son portadores de un cambio genético (aunque ellos mismos generalmente no padezcan la enfermedad podrían tener síntomas hemorrágicos leves) y transmiten este gene modificado a su hijo o hija. El síndrome de Bernard-Soulier afecta tanto a varones como a mujeres.
Los síntomas del síndrome de Bernard-Soulier varían de una persona a otra. Las señales del trastorno generalmente se notan por primera vez durante la infancia.
Las personas con síndrome de Bernard-Soulier pueden presentar los siguientes síntomas:
En la edad adulta temprana, el síndrome de Bernard-Soulier puede provocar más problemas en mujeres que en varones debido al riesgo de hemorragia relacionado con la menstruación y el parto.
No hay una sola prueba que pueda diagnosticar todos los trastornos de la función plaquetaria. El diagnóstico del síndrome de Bernard-Soulier requiere de un cuidadoso historial médico y de una serie de pruebas que debería realizar un especialista en un centro de tratamiento para trastornos de la coagulación.
En personas con síndrome de Bernard-Soulier:
Nota: Algunas pruebas no están disponibles en todos los centros.
El síndrome de Bernard-Soulier algunas veces es incorrectamente diagnosticado como trombocitopenia inmunitaria, un trastorno plaquetario adquirido en el cual el conteo plaquetario es bajo.
La mayoría de las personas que padece el síndrome de Bernard-Soulier requiere tratamiento antes de intervenciones quirúrgicas (incluso trabajos dentales) o después de lesiones o accidentes. Algunas personas necesitarán tratamiento para hemorragias nasales graves u otros síntomas hemorrágicos, tales como periodos menstruales abundantes.
En caso necesario, el síndrome de Bernard-Soulier puede recibir tratamiento con las siguientes opciones:
Para obtener más información consulte Productos de tratamiento.
Las personas que padecen el síndrome de Bernard-Soulier no deberían tomar aspirina (ácido acetilsalicílico), otros medicamentos antiinflamatorios no esteroides (AINE, tales como ibuprofeno y naproxeno), o anticoagulantes, ya que estos podrían empeorar los síntomas hemorrágicos.
La trombastenia de Glanzmann es un trastorno hereditario de la función plaquetaria causado por una anomalía en el receptor del fibrinógeno (también llamado receptor GPIIb/IIIa; véase la figura 2). Los receptores son proteínas en la superficie de las plaquetas que las ayudan a interactuar con otras células o sustancias sanguíneas y a responder ante ellas. Dado que el receptor del fibrinógeno está ausente o no funciona adecuadamente, las plaquetas no se adhieren entre sí en el lugar de la lesión. El resultado es que el tapón plaquetario no se forma normalmente.
La trombastenia de Glanzmann es un trastorno autosómico recesivo, lo que quiere decir que ambos padres son portadores de un cambio genético (aunque ellos mismos generalmente no padezcan la enfermedad) y transmiten este gene modificado a su hijo o hija. La trombastenia de Glanzmann afecta tanto a varones como a mujeres.
Los síntomas de la trombastenia de Glanzmann varían de una persona a otra y pueden ser desde leves hasta hemorragias que podrían poner en peligro la vida. Las señales del trastorno generalmente se notan por primera vez durante la infancia.
Las personas con trombastenia de Glanzmann pueden presentar los siguientes síntomas:
Durante la edad adulta temprana, la trombastenia de Glanzmann puede provocar más problemas en mujeres que en hombres debido al riesgo de hemorragia relacionado con la menstruación y el parto.
No hay una sola prueba que pueda diagnosticar todos los trastornos de la función plaquetaria. El diagnóstico de la trombastenia de Glanzmann requiere de un cuidadoso historial médico y de una serie de pruebas que debería realizar un especialista en un centro de tratamiento para trastornos de la coagulación.
En personas con trombastenia de Glanzmann:
Nota: Algunas pruebas no están disponibles en todos los centros.
La mayoría de las personas que padece trombastenia de Glanzmann requiere tratamiento antes de intervenciones quirúrgicas (incluso trabajos dentales) o después de lesiones o accidentes. Algunas personas necesitarán tratamiento para hemorragias nasales graves u otras hemorragias.
En caso necesario, la trombastenia de Glanzmann puede recibir tratamiento con las siguientes opciones:
Para obtener más información consulte Productos de tratamiento.
Las personas que padecen trombastenia de Glanzmann no deberían tomar aspirina, medicamentos antiinflamatorios no esteroides (AINE, tales como ibuprofeno y naproxeno) o anticoagulantes, ya que estos podrían empeorar los síntomas hemorrágicos.
Los gránulos plaquetarios son pequeños paquetes al interior de las plaquetas en los que se almacenan proteínas y otras sustancias químicas. El contenido de los gránulos se libera durante la fase de secreción de la activación plaquetaria (véase la figura 1), y funciona como señales químicas para reclutar más plaquetas y otras células al lugar de la lesión a fin de detener la hemorragia. Hay dos tipos de gránulos: gránulos alfa y gránulos densos (véase la figura 2).
Los trastornos de los gránulos plaquetarios constituyen un grupo diverso de trastornos hereditarios. Algunos son causados por una ausencia de gránulos o su contenido, pero los más comunes se deben a la incapacidad de las plaquetas para vaciar el contenido de los gránulos al torrente sanguíneo.
La manera en la que se heredan los trastornos de los gránulos plaquetarios (es decir, se transmiten de padres a hijos) es menos consistente que en otros tipos de trastornos plaquetarios y varía de una persona a otra.
Los síntomas de los trastornos de los gránulos plaquetarios varían de una persona a otra.
Las personas con trastornos de los gránulos plaquetarios podrían presentar los siguientes síntomas:
No hay una sola prueba que pueda diagnosticar todos los trastornos de la función plaquetaria. El diagnóstico de los trastornos de los gránulos plaquetarios requiere de un cuidadoso historial médico y de una serie de pruebas que debería realizar un especialista en un centro de tratamiento para trastornos de la coagulación.
En personas con trastornos de los gránulos plaquetarios:
Muchas personas con trastornos de los gránulos plaquetarios necesitan tratamiento antes de intervenciones quirúrgicas (incluso trabajos dentales) o después de lesiones o accidentes.
En caso necesario, las personas con trastornos de los gránulos plaquetarios pueden recibir tratamiento con las siguientes opciones:
Para obtener más información consulte Productos de tratamiento.
Las personas que padecen trastornos de los gránulos plaquetarios no deberían tomar aspirina (ácido acetilsalicílico), medicamentos antiinflamatorios no esteroides (AINE, tales como ibuprofeno y naproxeno) y anticoagulantes, ya que podrían empeorar sus síntomas hemorrágicos, a menos que un médico familiarizado con el trastorno se los recete con un fin específico.
Hay muchos otros tipos de trastornos hereditarios de la función plaquetaria; sin embargo, los principios de tratamiento siguen siendo los mismos. Algunos trastornos de la función plaquetaria podrían estar relacionados con otros trastornos sanguíneos. En casos particulares podría estar indicada la investigación de tales trastornos sanguíneos.
Todos los tratamientos pueden tener efectos secundarios. Las personas con trastornos plaquetarios hereditarios deberían hablar con su proveedor de atención médica sobre posibles efectos secundarios antes de tomar cualquier medicamento. Pregunte a su proveedor de atención médica si desea obtener más información sobre medicamentos y remedios herbales que no deberían ser usados por personas con trastornos de la coagulación. En raras circunstancias, estos medicamentos tendrían que ser considerados para el tratamiento de otros trastornos médicos. Su uso debería abordarse con el médico que encabeza el tratamiento del trastorno plaquetario.
Los fármacos antifibrinolíticos ácido tranexámico y ácido aminocaproico se utilizan para evitar la degradación de un coágulo en ciertas partes del cuerpo, tales como nariz, boca, vejiga y útero. Estos medicamentos son muy útiles en muchas situaciones; por ejemplo, para trabajos dentales, pero no son adecuados para hemorragias genitourinarias. Pueden utilizarse para reducir la hemorragia durante y después de algunos tipos de cirugías. Los fármacos antifibrinolíticos también se utilizan para ayudar a controlar el flujo menstrual abundante. Pueden aplicarse directamente en los sitios de hemorragias en la nariz o la boca. También pueden administrarse por vía oral o por inyección.
El factor VIIa recombinante es un factor de la coagulación activado que se administra mediante inyección. Puede ser eficaz para el tratamiento de hemorragias en algunos pacientes con trombastenia de Glanzmann o síndrome de Bernard-Soulier, así como para prevenir hemorragias antes de cirugías. Es particularmente útil como alternativa a la transfusión de plaquetas a fin de evitar que los pacientes desarrollen anticuerpos a las plaquetas, o para proporcionar tratamiento en casos en los que ya han aparecido anticuerpos (véase la sección ‘Transfusiones de plaquetas’ más abajo). El factor VIIa recombinante se fabrica en un laboratorio y no a partir de plasma humano.
La desmopresina (DDAVP) es una hormona sintética que puede ayudar a controlar hemorragias en casos de emergencia o durante cirugías. Puede administrarse por vía intravenosa, bajo la piel (por vía subcutánea) o como aerosol nasal. La desmopresina no funciona para todos los trastornos plaquetarios ni en todas las personas. Después de recibir desmopresina es importante restringir el consumo de líquidos durante las siguientes 24 horas. La desmopresina generalmente no se administra a personas con un mayor riesgo de enfermedad cardiovascular.
Los selladores de fibrina (también llamados goma de fibrina) están disponibles en algunos países. Pueden usarse para el tratamiento de lesiones externas y durante trabajos dentales tales como extracciones. Los selladores de fibrina se aplican directamente en el sito de la hemorragia.
La terapia de supresión hormonal (anticonceptivos) y el dispositivo/sistema intrauterino liberador de levonorgestrel pueden usarse para ayudar a controlar el sangrado menstrual abundante en mujeres que no desean embarazarse.
La reposición de hierro es necesaria para el tratamiento de la anemia o de la deficiencia de hierro sin anemia, causadas por hemorragias excesivas o prolongadas. Los suplementos de hierro pueden administrarse por vía oral o por inyección. Las personas que tienen hemorragias frecuentes deberían someterse a pruebas de sangre a fin de monitorear la anemia y/o bajas concentraciones de hierro.
Las transfusiones de plaquetas podrían ser necesarias para el tratamiento de hemorragias graves o de difícil control, así como para prevenir hemorragias durante o después de cirugías. Si bien las transfusiones de plaquetas pueden ser muy eficaces, se evitan en la medida de lo posible ya que algunas personas podrían desarrollar anticuerpos a las plaquetas transfundidas, lo cual reduciría la eficacia de futuras transfusiones. En algunos países se seleccionan las plaquetas a fin de reducir el riesgo de aparición de inhibidores, lo que se conoce como plaquetas seleccionadas con base en el antígeno leucocitario humano (HLA por su sigla en inglés).
Las mujeres con trastornos hereditarios de la función plaquetaria podrían presentar más síntomas que los hombres debido al riesgo de hemorragia relacionado con la menstruación y el parto. Las niñas podrían presentar hemorragia abundante cuando empiezan a menstruar.
Las mujeres con trastornos hereditarios de la función plaquetaria podrían presentar un flujo menstrual más abundante y/o prolongado, el cual podría generar deficiencia de hierro (bajas concentraciones de hierro, lo que provoca debilidad y cansancio) y/o anemia (bajas concentraciones de glóbulos rojos).
Las mujeres con trastornos hereditarios de la función plaquetaria deberían, con suficiente antelación a un embarazo planeado, recibir asesoría genética sobre los riesgos de tener un(a) hijo(a) afectado(a) por su trastorno, y deberían consultar a un obstetra tan pronto sospechen que están embarazadas. El obstetra debería trabajar en estrecha colaboración con el personal del centro de tratamiento de trastornos de la coagulación a fin de proporcionar la mejor atención posible durante el embarazo y el parto, y de minimizar posibles complicaciones tanto para la madre como para el recién nacido.
El principal riesgo relacionado con el embarazo y el parto es la hemorragia posparto. Los trastornos de la coagulación están relacionados con un mayor riesgo de hemorragia, tanto inmediatamente después del parto como varias semanas después del mismo. Por lo tanto, las mujeres con trastornos hereditarios de la función plaquetaria deberían colaborar con sus médicos (tanto con el hematólogo u otro especialista en trastornos de la coagulación como con el obstetra) a fin de preparar un plan de parto personalizado. Este plan debería abarcar todas las fases del trabajo de parto, incluyendo el parto de la placenta, a fin de reducir el riesgo y la gravedad de la hemorragia. El tratamiento será diferente para cada mujer y dependerá de su historial personal y familiar de síntomas hemorrágicos, del diagnóstico y de la gravedad del trastorno de la función plaquetaria, así como del tipo de parto. Las mujeres con trastornos de la función plaquetaria deberían consultar a su proveedor de atención médica inmediatamente si la hemorragia posparto fuera excesiva.
En algunas circunstancias, los bebés nacidos de mujeres con trastornos hereditarios de la función plaquetaria también podrían correr el riesgo de heredar el trastorno y presentar hemorragias. Debiera evitarse un trabajo de parto difícil y prolongado, al igual que alumbramientos con instrumentos como fórceps o ventosas.
Saber que usted o un miembro de su familia tiene un trastorno hereditario de la función plaquetaria puede ser perturbador y podría llegar a experimentar una gama de diferentes emociones. En algunas personas puede provocar miedo y ansiedad, mientras que para otras poder dar un nombre a los síntomas que han experimentado puede ser de gran alivio. Los padres podrían sentirse culpables al saber que su hijo(a) ha heredado un trastorno genético. Todos estos sentimientos son normales y es probable que cambien al paso del tiempo, conforme aprende más sobre el trastorno y el impacto que tendrá en su vida o la de los miembros de su familia.
Hablar con otras personas –amigos, padres, profesionales de la salud y otras personas con trastornos hereditarios de la función plaquetaria– puede resultar reconfortante. Aprender todo lo que sea posible acerca del trastorno le ayudará a sentirse más confiado y disipará sus miedos. Póngase en contacto con la organización de pacientes o centro de tratamiento de trastornos de la coagulación más cercanos al lugar donde vive para hacer preguntas y abordar sus opciones. Los grupos de pacientes y centros de tratamiento pueden ubicarse a través de la página Internet de la FMH en wfh.org/es/encuentre-apoyo-local/.
Las personas con trastornos hereditarios de la función plaquetaria deberían ser atendidas por un centro médico especializado en el diagnóstico y el tratamiento de trastornos de la coagulación, ya que es el que puede ofrecer la mejor atención e información.
Una dieta sana y ejercicio cotidiano ayudarán al cuerpo a mantenerse fuerte y saludable. El ejercicio también puede ayudar a disminuir el estrés, la ansiedad y la depresión, y reducir la frecuencia y la gravedad de los episodios hemorrágicos.
Algunos alimentos o aditivos alimentarios, tales como alcohol, aceites de pescado, el hongo chino oreja de árbol, el ajoene (un componente del ajo), y algunos otros remedios herbales pueden afectar la función plaquetaria y empeorar los síntomas.
Una buena higiene oral es indispensable para evitar caries y enfermedad periodontal (de las encías). Para las personas con trastornos hereditarios de la función plaquetaria mantener una buena salud dental es muy importante a fin de reducir la necesidad de cirugías dentales que podrían complicarse debido a hemorragias excesivas o prolongadas. Las personas con trastornos de la función plaquetaria deberían:
Procedimientos invasores tales como raspados, extracciones o endodoncias pueden provocar hemorragias en personas con trastornos de la función plaquetaria. El dentista debería consultar al centro de tratamiento de trastornos de la coagulación a fin de determinar el posible riesgo individual y establecer un plan adecuado para evitar o controlar hemorragias en cualquier procedimiento. Podría ser necesario tomar medicamentos antes de los procedimientos para evitar hemorragias y garantizar una intervención y una recuperación sin complicaciones.
Las personas con trastornos de la función plaquetaria deberían vacunarse. Podría ser necesario administrar las vacunas por vía subcutánea (bajo la piel), y no intramuscular, a fin de evitar el riesgo de hemorragia, y esto debería consultarse con el médico que se encarga del trastorno plaquetario.
Consulte a su médico antes de tomar medicamentos o remedios herbales. Deben evitarse algunos medicamentos de venta libre como la aspirina (ácido acetilsalicílico o ASA) o medicamentos antiinflamatorios no esteroides (AINE, como ibuprofeno y naproxeno) ya que interfieren con la función plaquetaria. Muchos otros medicamentos pueden también afectar la función plaquetaria, entre ellos medicamentos que adelgazan la sangre (anticoagulantes), algunos antibióticos, medicamentos para el corazón, antidepresivos, anestésicos y antihistamínicos. Los medicamentos que interfieren con la función plaquetaria no deberían usarse sin la recomendación específica de un médico que esté familiarizado con su trastorno plaquetario.
Las personas con trastornos de la coagulación siempre deberían llevar consigo información sobre su padecimiento, el tratamiento necesario, y el nombre y número telefónico de su médico o centro de tratamiento. En casos de emergencia, un brazalete médico u otra identificación como la tarjeta médica internacional de la FMH, ayudarán a notificar al personal de salud sobre su trastorno hereditario de la función plaquetaria. Antes de viajar obtenga la dirección y el número telefónico del centro de tratamiento de trastornos de la coagulación de su(s) lugar(es) de destino y lleve dicha información consigo en caso de que necesite atención. Los centros de tratamiento pueden ubicarse usando la página Internet de la FMH (wfh.org/es/encuentre-apoyo-local/).






