Les plaquettes sont de petites cellules en forme de disque qui circulent dans le sang. Elles jouent un rôle important dans la formation des caillots sanguins qui visent à arrêter les saignements et dans la réparation des vaisseaux sanguins endommagés.
Lorsqu’un vaisseau sanguin est endommagé, les plaquettes commencent à arrêter le saignement en formant ce que l’on appelle un clou ou un bouchon plaquettaire. Ce processus se déroule en trois étapes (voir figure 1) :
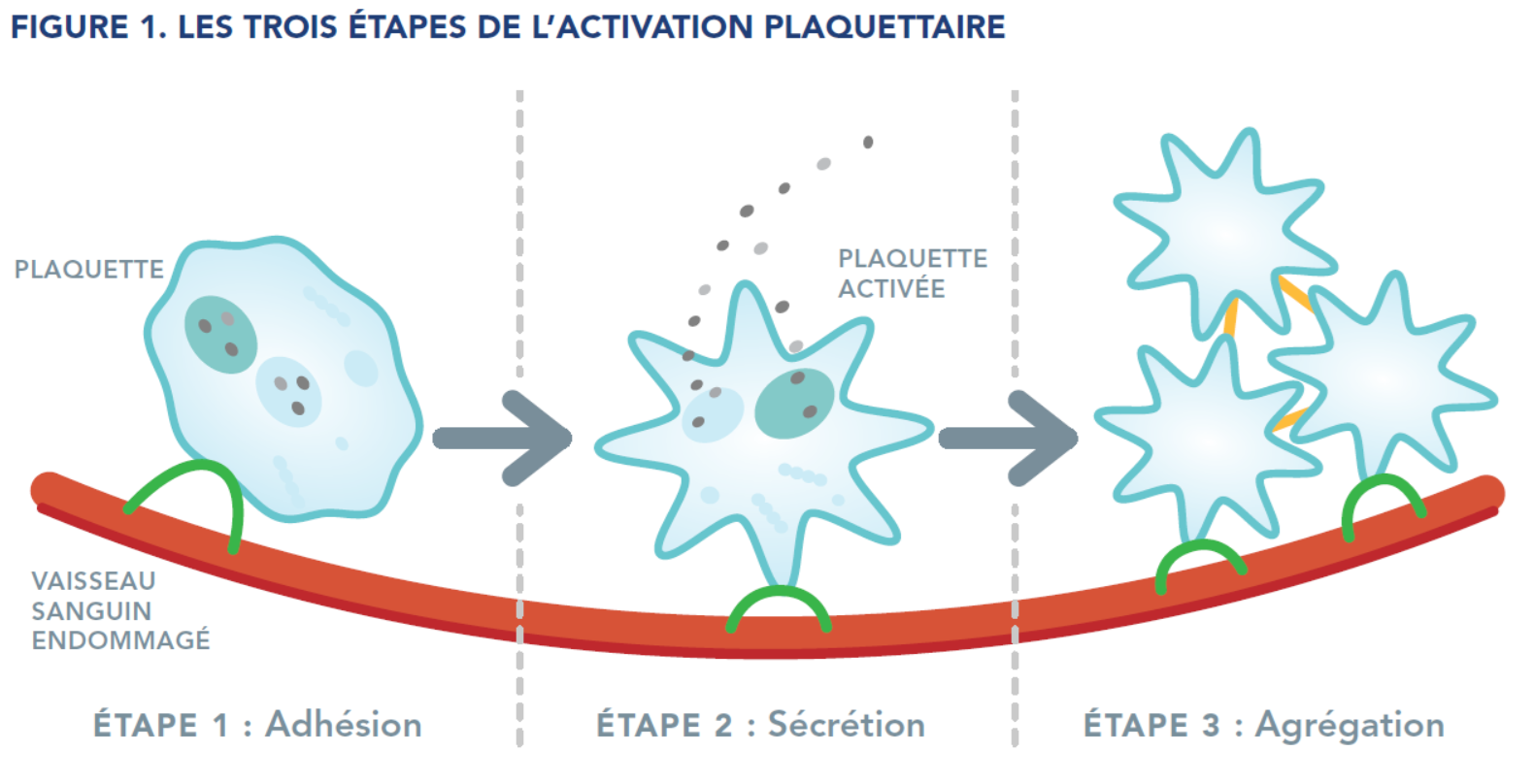
Pendant la formation du clou plaquettaire, des protéines appelées facteurs de coagulation sont également acheminées vers le site de la blessure. Ces facteurs de coagulation agissent ensemble à la surface des plaquettes pour renforcer le clou plaquettaire en formant un maillage appelé caillot de fibrine.
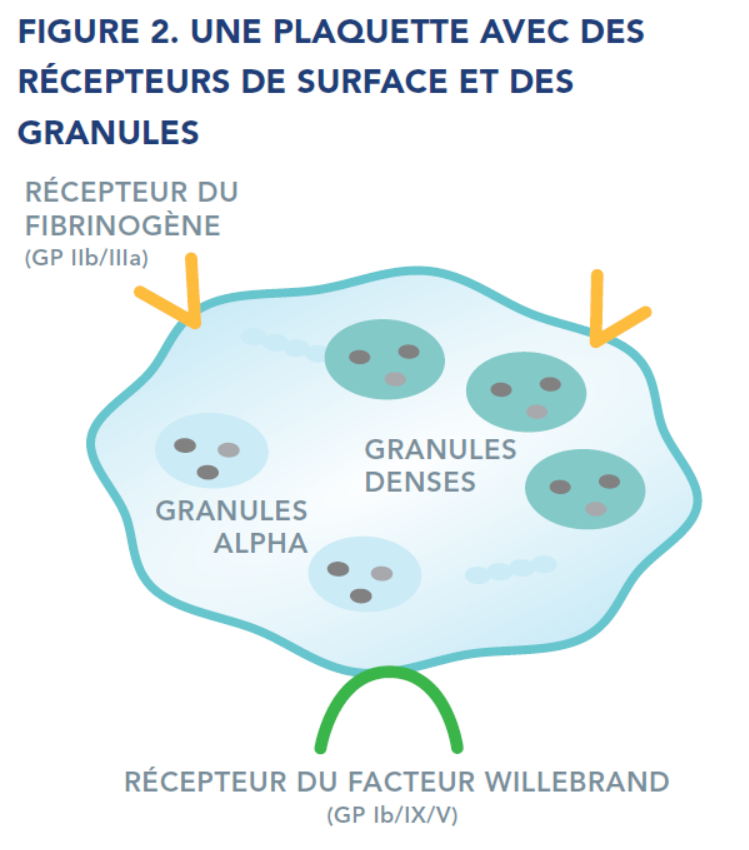
Les plaquettes sont composées de plusieurs éléments, tels que des récepteurs à leur surface et des granules internes, qui contribuent tous à la formation du caillot.
Récepteurs
Les récepteurs sont des protéines présentes à la surface des plaquettes qui leur permettent d’interagir entre elles, et de répondre aux sollicitations de substances chimiques, de protéines, ainsi que d’autres cellules présentes dans le sang.
Granules
Les granules sont de petites poches situées à l’intérieur des plaquettes où sont stockées des protéines et certaines substances chimiques. Le contenu des granules est libéré par les plaquettes pendant la phase de sécrétion lors de l’activation plaquettaire. Ces molécules agissent comme des signaux chimiques pour attirer davantage de plaquettes et d’autres cellules sur le site de la blessure afin d’arrêter le saignement.
Il existe deux types de granules : les granules alpha et les granules denses. Leur contenu diffère. Chaque catégorie agit à sa façon pour attirer davantage de plaquettes, activer les facteurs de coagulation et arrêter le saignement.
Les dysfonctions plaquettaires sont des troubles caractérisés par une anomalie de la fonction plaquettaire. Le clou plaquettaire ne se formant pas correctement, les saignements peuvent durer plus longtemps que la normale. Les personnes souffrant de dysfonctions plaquettaires ont tendance à faire des ecchymoses ou à saigner plus facilement que la normale. Les dysfonctions plaquettaires peuvent être causées par un problème au niveau de l’un des récepteurs, des granules ou des processus d’activation à l’intérieur des plaquettes, et peuvent ne pas être liées à une faible numération plaquettaire.
Les dysfonctions plaquettaires sont héréditaires (les parents les transmettent à leurs enfants) ou acquises. La présente brochure porte sur les dysfonctions plaquettaires héréditaires.
Le syndrome de Bernard-Soulier est une dysfonction plaquettaire causée par une anomalie du récepteur du facteur Willebrand (voir figure 2). Ce récepteur est également appelé glycoprotéine (GP) Ib/IX/V. Les récepteurs sont des protéines présentes à la surface des plaquettes qui les aident à interagir entre elles, ainsi qu’avec d’autres cellules ou substances sanguines. Le récepteur du facteur Willebrand étant absent ou ne fonctionnant pas correctement, les plaquettes ne peuvent pas se lier au facteur Willebrand et n’adhèrent pas à la paroi du vaisseau sanguin endommagé comme elles devraient le faire. Par conséquent, le clou plaquettaire ne se forme pas normalement.
Le syndrome de Bernard-Soulier est une maladie autosomique récessive, ce qui signifie que les deux parents sont porteurs d’une mutation génétique (même s’ils ne sont généralement pas eux-mêmes atteints de la maladie, ils peuvent présenter des symptômes hémorragiques légers) et qu’ils transmettent ce gène modifié à leur enfant. Le syndrome de Bernard-Soulier touche aussi bien les hommes que les femmes.
Les symptômes du syndrome de Bernard-Soulier varient d’une personne à l’autre. Les signes de la maladie sont généralement observés dès la petite enfance.
Les personnes atteintes du syndrome de Bernard-Soulier peuvent présenter les symptômes suivants :
Le syndrome de Bernard-Soulier peut s’avérer plus problématique pour les femmes que pour les hommes à la puberté et dans les années qui suivent, en raison du risque de saignement associé à la menstruation et à l’accouchement.
Il n’existe pas de test unique pour diagnostiquer l’ensemble des dysfonctions plaquettaires. Pour diagnostiquer le syndrome de Bernard-Soulier, il convient de procéder à un examen minutieux des antécédents médicaux et à une batterie de tests en laboratoire qui devraient être réalisés dans un centre spécialisé dans les troubles de la coagulation.
Chez les personnes atteintes du syndrome de Bernard-Soulier :
Remarque : certains tests ne sont pas disponibles dans tous les centres.
Le syndrome de Bernard-Soulier est parfois confondu avec une thrombocytopénie immune, dysfonction plaquettaire acquise qui se caractérise par une numération plaquettaire basse.
La plupart des personnes atteintes du syndrome de Bernard-Soulier doivent être prises en charge avant toute intervention chirurgicale (notamment les soins dentaires) ou après une blessure ou un accident. Certaines personnes auront besoin d’une prise en charge médicamenteuse en cas de saignements de nez importants ou d’autres symptômes hémorragiques, tels que des saignements menstruels abondants.
En cas de besoin, le syndrome de Bernard-Soulier peut être traité avec :
Voir « Traitements » pour plus de détails.
Toute prise d’aspirine (acide acétylsalicylique), d’anti-inflammatoire non stéroïdien (ibuprofène ou naproxène, par exemple) ou d’anticoagulant est strictement déconseillée, car elle peut renforcer les saignements.
La thrombasthénie de Glanzmann est une dysfonction plaquettaire héréditaire causée par une anomalie du récepteur du fibrinogène (également appelé GP IIb/IIIa, voir figure 2). Les récepteurs sont des protéines présentes à la surface des plaquettes qui les aident à interagir entre elles, ainsi qu’avec d’autres cellules ou substances sanguines. Le récepteur du fibrinogène étant absent ou ne fonctionnant pas correctement, les plaquettes n’adhèrent pas les unes aux autres à l’endroit de la lésion. Par conséquent, le clou plaquettaire ne se forme pas normalement.
La thrombasthénie de Glanzmann est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs de la mutation génétique (même si normalement ils ne sont pas eux-mêmes atteints de la maladie) pour la transmettre à leur enfant. La thrombasthénie de Glanzmann touche aussi bien les hommes que les femmes.
Les symptômes de la thrombasthénie de Glanzmann varient d’une personne à l’autre. Les saignements peuvent être bénins, mais peuvent aussi engager le pronostic vital. Les signes de la maladie sont généralement observés dès la petite enfance.
Les personnes atteintes de la thrombasthénie de Glanzmann peuvent présenter les symptômes suivants :
La thrombasthénie de Glanzmann peut s’avérer plus problématique pour les femmes que pour les hommes à la puberté et dans les années qui suivent, en raison du risque de saignement associé à la menstruation et à l’accouchement.
Il n’existe pas de test unique pour diagnostiquer l’ensemble des dysfonctions plaquettaires. Pour diagnostiquer la thrombasthénie de Glanzmann, il convient de procéder à un examen minutieux des antécédents médicaux et à une batterie de tests en laboratoire qui devraient être réalisés dans un centre spécialisé dans les troubles de la coagulation.
Chez les personnes atteintes de la thrombasthénie de Glanzmann :
Remarque : certains tests ne sont pas disponibles dans tous les centres.
La plupart des personnes atteintes de la thrombasthénie de Glanzmann doivent être prises en charge avant toute intervention chirurgicale (notamment les soins dentaires) ou après une blessure ou un accident. Certaines personnes auront besoin d’une prise en charge médicamenteuse en cas de saignements de nez importants ou d’autres symptômes hémorragiques.
En cas de besoin, la thrombasthénie de Glanzmann peut être traitée avec :
Voir « Traitements » pour plus de détails.
Toute prise d’aspirine (acide acétylsalicylique), d’anti-inflammatoire non stéroïdien (ibuprofène ou naproxène, par exemple) ou d’anticoagulant est strictement déconseillée, car elle peut renforcer les saignements.
Les granules sont de petites poches situées à l’intérieur des plaquettes où sont stockées des protéines et certaines substances chimiques. Le contenu des granules est libéré par les plaquettes pendant la phase de sécrétion lors de l’activation plaquettaire (voir figure 1). Ces molécules agissent comme des signaux chimiques pour attirer davantage de plaquettes et d’autres cellules sur le site de la blessure afin d’arrêter le saignement. Il existe deux types de granules : les granules alpha et les granules denses (voir figure 2).
Les dysfonctions de la sécrétion plaquettaire sont des troubles hétérogènes. Certaines sont dues à une absence de granules ou de leur contenu, mais les plus communes s’expliquent par une incapacité des plaquettes à libérer leur contenu dans le flux sanguin.
Le mode de transmission (des parents à l’enfant) des dysfonctions de la sécrétion plaquettaire varie d’un trouble à l’autre et d’une personne à l’autre.
Les symptômes associés aux dysfonctions de la sécrétion plaquettaire varient d’une personne à l’autre.
Les personnes atteintes d’une dysfonction de la sécrétion plaquettaire peuvent présenter les symptômes suivants :
Il n’existe pas de test unique pour diagnostiquer l’ensemble des dysfonctions plaquettaires. Pour diagnostiquer les dysfonctions de la sécrétion plaquettaire, il convient de procéder à un examen minutieux des antécédents médicaux et à une batterie de tests en laboratoire qui devraient être réalisés dans un centre spécialisé dans les troubles de la coagulation.
Chez les personnes atteintes de dysfonctions de la sécrétion plaquettaire :
La plupart des personnes atteintes de dysfonctions de la sécrétion plaquettaire doivent être prises en charge avant toute intervention chirurgicale (notamment les soins dentaires) ou après une blessure ou un accident.
En cas de besoin, les dysfonctions de la sécrétion plaquettaire peuvent être traitées avec :
Voir « Traitements » pour plus de détails.
Sauf prescription pour un motif bien précis par un médecin spécialisé dans ce type de dysfonctions, toute prise d’aspirine (acide acétylsalicylique), d’anti-inflammatoire non stéroïdien (ibuprofène ou naproxène, par exemple) ou d’anticoagulant est strictement déconseillée, car elle peut renforcer les saignements.
Il existe de nombreuses autres dysfonctions plaquettaires héréditaires, mais les principes de prise en charge restent les mêmes. Certaines dysfonctions plaquettaires peuvent être associées à d’autres maladies du sang. Dans certains cas, la recherche d’autres troubles sanguins peut être recommandée.
Tout traitement peut avoir des effets secondaires. Les personnes atteintes de dysfonctions plaquettaires sont invitées à consulter un professionnel de santé sur les éventuels effets indésirables avant toute prise de médicament. Pour plus d’information sur les médicaments et les traitements à base de plantes qui ne doivent pas être utilisés par les personnes atteintes de troubles de la coagulation, veuillez vous adresser au professionnel de santé qui vous suit. Dans de rares circonstances, ces médicaments peuvent être envisagés pour la prise en charge d’autres pathologies. Il convient de discuter avec le médecin responsable du suivi de la dysfonction plaquettaire du recours à de tels traitements.
Les agents anti-fibrinolytiques, comme l’acide tranexamique et l’acide aminocaproïque, sont utilisés pour stabiliser le caillot formé dans certaines parties du corps, notamment le nez, la bouche, la vessie et l’utérus. Ces médicaments sont utiles dans de nombreuses situations, par exemple au moment de procéder à des soins dentaires, mais ne sont pas recommandés en cas d’hémorragie génito-urinaire. Ils peuvent être utilisés pour réduire les saignements pendant et après certains types d’interventions chirurgicales. Les agents anti-fibrinolytiques sont également utilisés pour gérer les saignements menstruels abondants. Ils peuvent être appliqués directement sur les sites de saignement, dans le nez ou la bouche. Ils peuvent également être administrés par voie orale ou par injection.
Le facteur VIIa recombinant est un facteur de coagulation activé qui est administré par injection. Il peut être efficace pour traiter les saignements chez certaines personnes atteintes de thrombasthénie de Glanzmann ou du syndrome de Bernard-Soulier et pour prévenir les saignements avant une intervention chirurgicale. Il constitue une alternative particulièrement utile à la transfusion de plaquettes pour éviter tout développement d’anticorps dirigés contre les plaquettes ou pour traiter les patients ayant développé des anticorps (voir plus bas « Transfusions de plaquettes »). Le facteur VIIa recombinant est fabriqué en laboratoire et non à partir de plasma humain.
La desmopressine (DDAVP) est une hormone synthétique qui peut aider à maîtriser un saignement en cas d’urgence ou pendant une intervention chirurgicale. Elle peut être injectée par voie intraveineuse ou sous la peau (sous-cutanée), ou administrée sous forme de pulvérisation nasale. La desmopressine n’est pas efficace pour toutes les dysfonctions plaquettaires, ni chez tous les individus. Il est important de limiter la consommation de liquides dans les 24 heures suivant la prise de desmopressine. La desmopressine n’est généralement pas administrée aux personnes présentant un risque accru de maladie cardiovasculaire.
Les colles de fibrine sont disponibles dans certains pays. Elles peuvent être utilisées pour traiter certaines plaies externes et lors de procédures dentaires, comme l’extraction d’une dent. Les colles de fibrine sont appliquées directement sur le site de l’hémorragie.
Les femmes ne souhaitant pas avoir d’enfant peuvent opter pour un traitement hormonal suppressif (médicaments contraceptifs) ou un dispositif intra-utérin à libération de lévonorgestrel afin de gérer les saignements menstruels abondants.
Il convient de traiter par la prise d’un supplément de fer toute anémie ou carence en fer sans anémie causée par des saignements excessifs ou prolongés. Le traitement peut être administré par voie orale ou par injection. Les personnes qui saignent fréquemment doivent faire des analyses de sang pour surveiller toute anémie et/ou carence en fer.
Il peut être nécessaire de traiter des hémorragies graves ou incontrôlées ou de prévenir les hémorragies pendant ou après une intervention chirurgicale en procédant à une transfusion de plaquettes. Une telle transfusion est certes efficace, mais elle doit être, dans la mesure du possible, évitée pour ne pas développer d’anticorps dirigés contre les plaquettes transfusées, ce qui réduirait l’efficacité des transfusions suivantes. Dans certains pays, les plaquettes sont sélectionnées pour réduire le risque de survenue d’anticorps (en fonction de l’antigène leucocytaire humain).
Les femmes atteintes de dysfonctions plaquettaires héréditaires peuvent présenter davantage de symptômes que les hommes en raison du risque de saignement associé à la menstruation et à l’accouchement. Les filles peuvent avoir des saignements abondants à la puberté.
Les femmes atteintes de dysfonctions plaquettaires héréditaires peuvent avoir un flux menstruel plus abondant et/ou plus long, ce qui peut entraîner une carence en fer (faible taux de fer, ce qui entraîne un état de faiblesse et de la fatigue) et/ou une anémie (faible taux de globules rouges).
Avant toute grossesse, les femmes atteintes de dysfonctions plaquettaires héréditaires doivent bénéficier d’un conseil génétique sur les risques de donner naissance à un enfant atteint du même trouble. Il convient de consulter un obstétricien dès qu’elles pensent être enceintes. L’obstétricien devrait collaborer avec le personnel du centre de traitement des troubles de la coagulation afin d’assurer une prise en charge optimale au cours de la grossesse et de l’accouchement et de réduire les complications potentielles pour la mère et le nouveau-né.
Le principal risque lié à la grossesse et à l’accouchement est l’hémorragie post-partum. Les troubles de la coagulation sont associés à un risque accru de saignement, à la fois juste après, mais également pendant les semaines qui suivent l’accouchement. Les femmes atteintes de dysfonctions plaquettaires doivent donc discuter avec leurs médecins (spécialiste des troubles de la coagulation et obstétricien) afin d’élaborer un plan d’accouchement individuel. Ce plan doit couvrir toutes les étapes de l’accouchement, notamment la délivrance du placenta, afin de réduire le risque et la gravité des saignements. Le traitement varie pour chaque femme et dépend de ses antécédents personnels et familiaux, des symptômes hémorragiques, du diagnostic et de la gravité de la dysfonction plaquettaire, ainsi que du mode d’accouchement. Il est fortement recommandé aux femmes atteintes de dysfonctions plaquettaires de contacter immédiatement leur professionnel de santé si les saignements post-partum sont excessifs.
Dans certains cas, les enfants nés de femmes atteintes de dysfonctions plaquettaires héréditaires risquent également d’hériter du trouble et de présenter des saignements. Lors de l’accouchement, le travail difficile et prolongé, ainsi que le recours à des instruments comme les forceps ou l’extraction par aspiration, doivent être évités.
L’annonce du diagnostic d’une dysfonction plaquettaire héréditaire peut être bouleversante et vous pouvez ressentir tout un éventail d’émotions différentes. Certaines personnes peuvent ressentir de la peur et de l’anxiété, alors que pour d’autres, le fait de pouvoir mettre un nom sur les symptômes qu’elles ont ressentis peut s’avérer un énorme soulagement. Les parents peuvent se sentir coupables d’apprendre que leur enfant a hérité d’une maladie génétique. Tous ces sentiments sont normaux et sont susceptibles d’évoluer au fil du temps, à mesure que vous en apprendrez davantage sur la maladie et sur l’incidence qu’elle aura sur votre vie ou celle d’un membre de votre famille.
Il peut être réconfortant de parler avec d’autres personnes (amis, parents, professionnels de santé ou autres personnes atteintes de dysfonctions plaquettaires héréditaires). Plus vous en saurez sur la dysfonction plaquettaire concernée, plus vous serez confiant et rassuré. Prenez contact avec l’association de patients ou le centre de traitement des troubles de la coagulation de votre région pour poser toutes vos questions et discuter des options qui s’offrent à vous. Vous pouvez trouver les groupes de patients et les centres de traitement près de chez vous sur le site Internet de la Fédération mondiale de l’hémophilie à l’adresse suivante : wfh.org/fr/soutien-sur-le-plan-local/.
Les personnes atteintes de dysfonctions plaquettaires héréditaires doivent être suivies par un centre de traitement spécialisé dans le diagnostic et la prise en charge des troubles de la coagulation. C’est là que vous bénéficierez de la meilleure prise en charge et que vous obtiendrez les informations les plus pertinentes.
Une alimentation saine et une activité physique régulière permettent de conserver un corps sain et fort. L’exercice peut également contribuer à réduire le stress, l’anxiété et la dépression, ainsi que la fréquence et la gravité des saignements.
Certains aliments ou additifs alimentaires, comme l’alcool, les huiles de poisson, le champignon noir chinois, l’ajoène (un composant de l’ail), ainsi que plusieurs remèdes à base de plantes, peuvent affecter la fonction plaquettaire et aggraver les symptômes.
Une bonne hygiène bucco-dentaire est essentielle pour prévenir les caries et les maladies gingivales. Pour les personnes atteintes de dysfonctions plaquettaires héréditaires, il est très important de maintenir une bonne hygiène dentaire afin de réduire la nécessité de procédures dentaires, qui peuvent être compliquées par des saignements gingivaux excessifs ou prolongés. Les personnes atteintes de dysfonctions plaquettaires héréditaires doivent :
Les procédures invasives, comme le détartrage, les extractions ou la dévitalisation, peuvent provoquer des saignements chez les personnes atteintes de dysfonctions plaquettaires. Le dentiste doit consulter le centre de traitement des troubles de la coagulation afin de déterminer tout risque potentiel et d’élaborer un plan approprié pour prévenir ou traiter les saignements lors d’une quelconque procédure. Avant toute intervention, il peut être nécessaire de prendre des médicaments afin de prévenir les saignements et d’assurer une intervention et un rétablissement sans complications.
Les personnes atteintes de dysfonctions plaquettaires doivent être vaccinées. Il peut être nécessaire d’administrer les vaccins par voie sous-cutanée (sous la peau) plutôt que par voie intramusculaire afin d’éviter tout risque de saignement associé. Il convient d’en discuter avec le professionnel de santé spécialisé dans la dysfonction plaquettaire correspondante.
Consultez votre médecin avant toute prise de médicaments ou de traitements à base de plantes. Certains médicaments en vente libre, comme l’aspirine (acide acétylsalicylique) ou d’autres anti-inflammatoires non stéroïdiens (ibuprofène ou naproxène, par exemple), doivent être évités, car ils interfèrent avec la fonction plaquettaire. De nombreux autres médicaments peuvent également affecter la fonction plaquettaire, notamment les anticoagulants, certains antibiotiques, les médicaments pour le cœur, les antidépresseurs, les anesthésiques et les antihistaminiques. Les médicaments qui interfèrent avec la fonction plaquettaire ne doivent pas être utilisés sans l’avis spécifique d’un médecin connaissant bien votre dysfonction plaquettaire.
Les personnes atteintes de troubles de la coagulation doivent toujours avoir sur elles des informations sur leur maladie, le traitement nécessaire, ainsi que le nom et le numéro de téléphone de leur médecin ou de leur centre de traitement. En cas d’urgence, un bracelet d’alerte médicale ou tout autre moyen d’identification, tel que la carte médicale internationale de la FMH, informe le personnel de santé de votre dysfonction plaquettaire héréditaire. Avant de partir en voyage, trouvez l’adresse et le numéro de téléphone des centres de traitement des troubles de la coagulation à destination et conservez ces informations sur vous au cas où vous en auriez besoin. Les coordonnées des centres de traitement sont disponibles sur le site Internet de la FMH (wfh.org/fr/soutien-sur-le-plan-local/).



