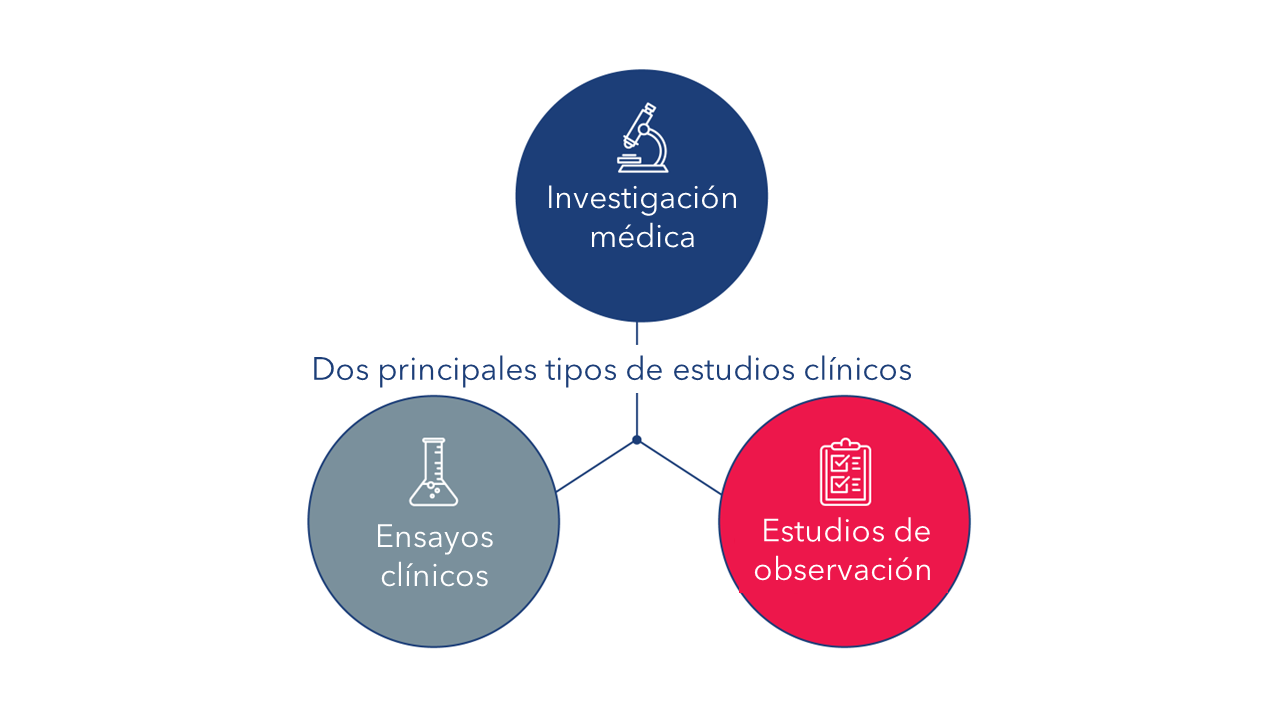
Los estudios clínicos generalmente involucran a seres humanos voluntarios, participantes o muestras (de sangre u otros tejidos) de seres humanos, con el propósito de incrementar los conocimientos médicos. Hay dos tipos principales de estudios clínicos: Ensayos clínicos (también llamados estudios de intervención) y estudios de observación.
Un ensayo clínico es un tipo de estudio de investigación en el que los investigadores prueban nuevas maneras de prevenir o detectar una enfermedad, o de administrar tratamiento para la misma. Los participantes en un ensayo clínico reciben intervenciones específicas de acuerdo con un protocolo detallado para dicho ensayo clínico. Tales intervenciones pueden ser productos médicos, como fármacos o dispositivos; procedimientos quirúrgicos; o cambios de comportamiento, como la dieta de un participante.
Los ensayos clínicos pueden comparar un nuevo método de tratamiento con uno habitual que ya se encuentre disponible, con un placebo o con la ausencia de intervención. Los ensayos clínicos son el principal método usado por los investigadores a fin de averiguar si un nuevo tratamiento es seguro y eficaz. Para que un fármaco se convierta en un medicamento que los médicos puedan recetar, primero debe probarse en una serie de ensayos clínicos llamados de Fase 1, Fase 2 y Fase 3. Después de los ensayos clínicos de Fase 3, los datos de los mismos se presentan a una agencia reguladora que entonces determina si el uso del fármaco debiera aprobarse.
Hay diferentes tipos de ensayos clínicos:
Los siguientes términos se usan con frecuencia para describir ensayos clínicos:
Un estudio de observación (también estudio observacional) es un tipo de estudio de investigación en el que los investigadores observan los efectos de una intervención en un grupo de participantes, sin intervenir. El investigador no asigna intervenciones específicas a los participantes, como en el ensayo clínico, pero los participantes podrían ya estar recibiendo un tratamiento que forme parte de su atención médica habitual. Los investigadores pueden entonces valorar las relaciones entre intervenciones y resultados de salud en las personas que reciben el tratamiento como parte de su atención habitual. Tales hallazgos podrían conducir a mayores investigaciones a través de un ensayo clínico. Existen diferentes tipos de estudios de observación. Un registro (padrón, censo) de pacientes es un tipo de estudio de observación.
Un registro de pacientes es un sistema organizado que utiliza métodos de un estudio de observación para recolectar datos sobre tratamientos, resultados clínicos y bienestar de una población definida por una enfermedad, trastorno o exposición específicos.
Para la comunidad de hemofilia, los siguientes son dos importantes registros de pacientes:
1. Registro Mundial de Trastornos de la Coagulación
El Registro Mundial de Trastornos de la Coagulación (RMTC) es un sistema de ingreso de datos basado en internet que ofrece una plataforma a una red de centros de tratamiento de hemofilia en todo el mundo con el propósito de recolectar datos uniformes y estandarizados de pacientes, y de orientar el ejercicio de la medicina. Con el consentimiento informado del paciente, el RMTC almacena datos sobre su enfermedad, pero que no permiten su identificación personal, tales como tipo y gravedad de la hemofilia, síntomas, tratamiento y resultados de salud.
Los profesionales de la salud que participan en el RMTC pueden usarlo para monitorear y dar seguimiento a la evolución de sus pacientes, y orientar su atención médica. Estos datos confidenciales y que no permiten la identificación personal también pueden usarse para ayudar a los investigadores a responder importantes preguntas sobre disparidades en la atención a escala mundial, así como ayudar a impulsar iniciativas de cabildeo/políticas de salud.
Haga clic aquí para visitar la página internet del RMTC.
2. Registro de Terapia Génica de la Federación Mundial de Hemofilia
Mediante un esfuerzo de colaboración internacional, la FMH creó un registro mundial de pacientes que reciben terapia génica, llamado Registro de Terapia Génica (RTG) de la FMH. El objetivo del RTG es ofrecer una base de datos sólida y con validez científica a todos los proveedores de atención médica que atienden a personas con hemofilia (PCH) que reciben terapia génica en cualquier lugar del mundo. Los datos recolectados a través del RTG de la FMH se utilizarán para valorar la seguridad y eficacia a largo plazo de la terapia génica en PCH.
Haga clic aquí para visitar el RTG de la FMH.
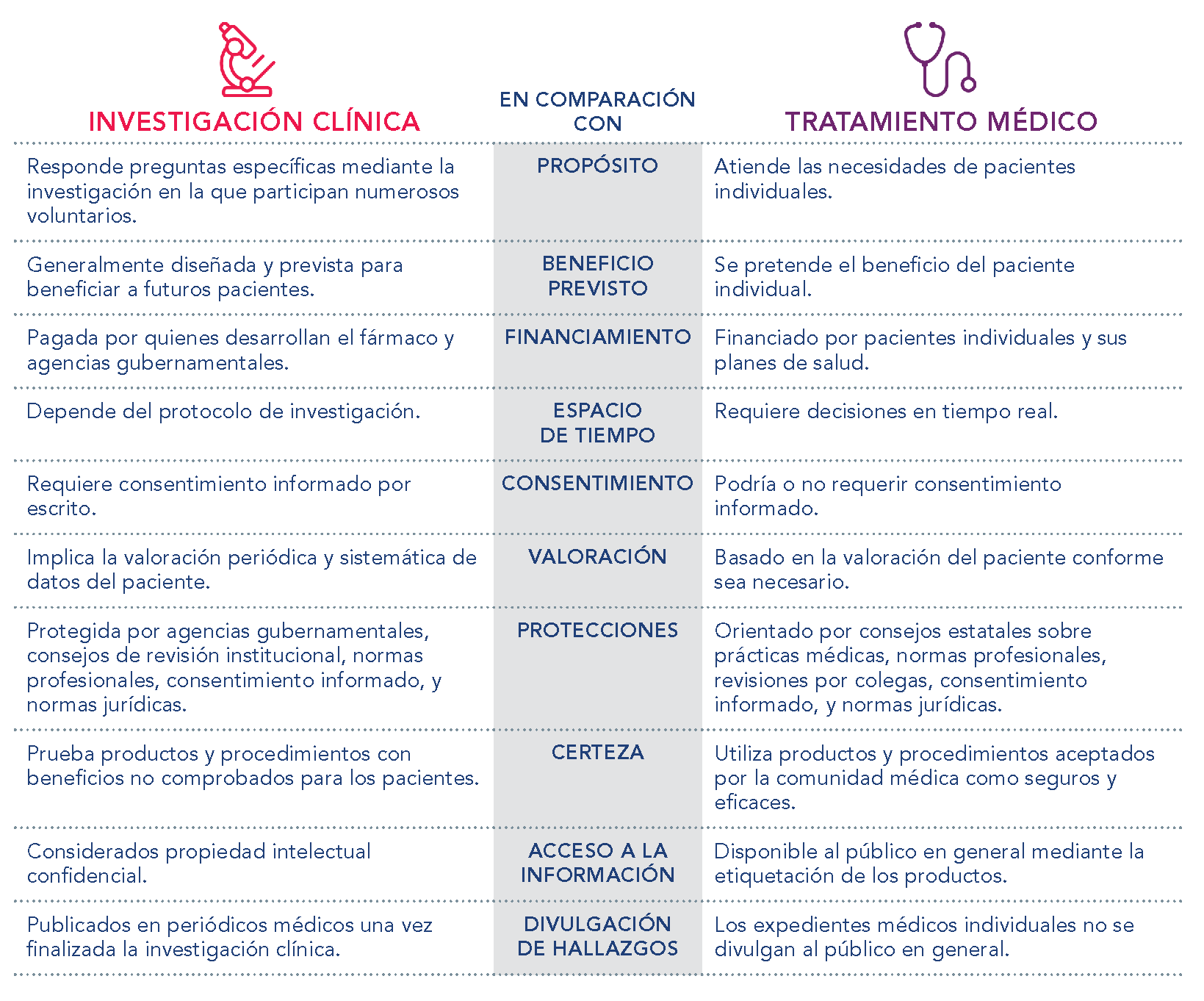
El siguiente cuadro puede resultar útil para comprender las diferencias entre la investigación clínica y un tratamiento médico.
Para que un fármaco se convierta en un tratamiento que los médicos puedan recetar, primero debe probarse en una serie de fases del ensayo clínico, y enseguida evaluarse y aprobarse por una agencia reguladora. Cada fase del ensayo clínico tiene un propósito, y estas avanzan en orden de la fase 1 a la fase 4.
Fase 1:
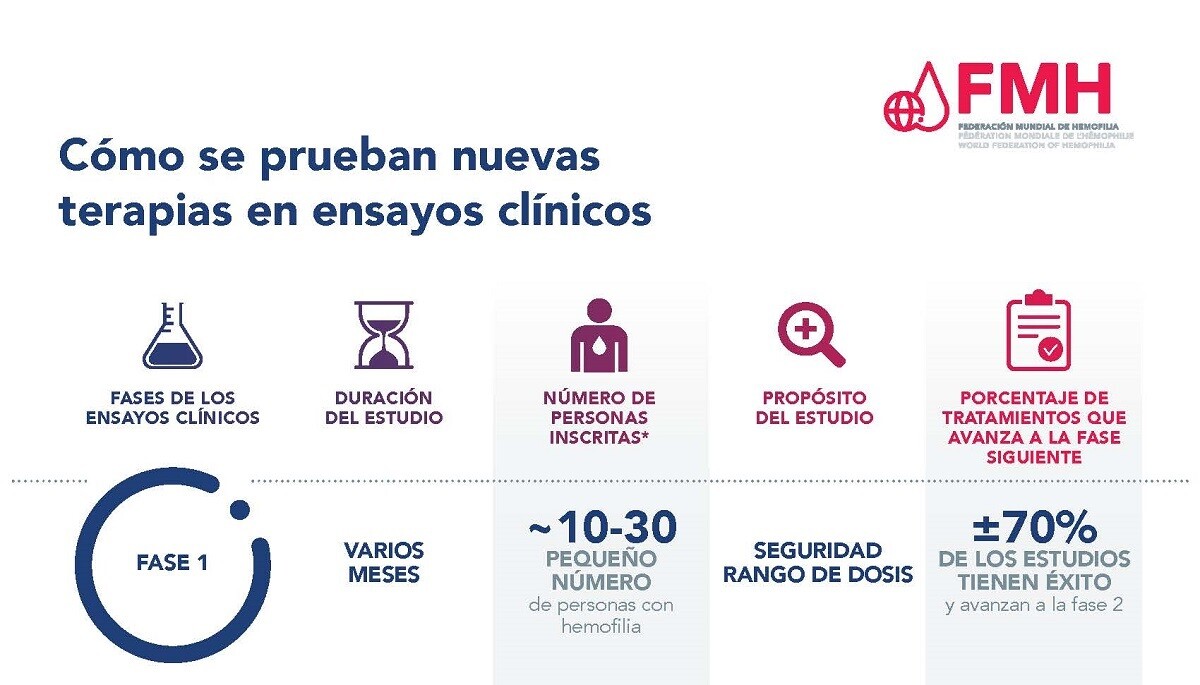
Esta es la primera vez que un nuevo tratamiento experimental se prueba en personas. El propósito de la fase 1 de un ensayo clínico es valorar la seguridad y cualesquiera efectos secundarios relacionados con el tratamiento. A menudo se realiza en un pequeño número de voluntarios sanos. Sin embargo, en el caso de enfermedades poco comunes, como la hemofilia, los ensayos de fase 1 generalmente se realizan en PCH. Los ensayos clínicos de fase 1 por lo general tardan varios meses, y aproximadamente el 70% de las terapias probadas en la fase 1 avanzan a la siguiente fase. El desarrollo clínico no va más allá de la fase 1 en el caso del 30% restante de las terapias que no demuestran su seguridad en la fase 1.
En los ensayos clínicos de fase 1 de terapia génica para la hemofilia se inscriben entre 10 y 30 personas con hemofilia.
Fase 2:
El propósito de la fase 2 de los ensayos clínicos es continuar probando la seguridad y los efectos secundarios relacionados con el tratamiento, pero también probar la eficacia y determinar la dosificación más eficaz. Los ensayos de fase 2 generalmente se realizan en más participantes que los ensayos de fase 1, y abarcan a personas que padecen la enfermedad. En algunas situaciones, los ensayos de fase 1 y de fase 2 se combinan y se conocen como ensayos de fase 1/2. Los ensayos clínicos de fase 2 por lo general tardan de varios meses hasta 2 años para realizarse. Se calcula que 33% de las terapias probadas en fase 2 avanzan a la siguiente fase. El desarrollo clínico no va más allá de la fase 2 si el tratamiento no demostrara ser eficaz o si hubiera preocupaciones de seguridad.
En los ensayos clínicos de fase 2 de terapia génica para la hemofilia se inscriben entre 10 y 50 personas con hemofilia. Esta cifra es menor que en el caso de otras enfermedades porque la hemofilia es un trastorno poco común. Los ensayos de fase 1 y de fase 2 a menudo se combinan en el caso de enfermedades poco comunes como la hemofilia. Al combinar las fases de un ensayo, los fabricantes de los fármacos esperan aceleran el calendario de desarrollo de nuevos medicamentos.
Fase 3:
El propósito de los ensayos clínicos de fase 3 es confirmar la eficacia del tratamiento, monitorear los efectos secundarios, y comparar el nuevo tratamiento con tratamientos habituales o similares. Los ensayos de fase 3, algunas veces llamados ‘ensayos trascendentales’, se realizan en un gran número de personas que padecen la enfermedad y en muchos lugares diferentes (nacionales e internacionales, dependiendo del estudio). Los ensayos clínicos de fase 3 algunas veces son aleatorizados y con frecuencia son de enmascaramiento doble. Los ensayos clínicos de fase 3 generalmente tiene una duración de 1 a 4 años y constituyen la última etapa antes de someter una solicitud para su aprobación a una agencia reguladora. Se calcula que un 25% de los ensayos de fase 3 avanzan a la fase 4.
Los datos del estudio trascendental de fase 3, y algunas veces datos de los ensayos clínicos de fase 1 y 2, se presentan a la agencia reguladora para su revisión. La agencia reguladora realiza análisis independientes sobre la seguridad y eficacia del tratamiento y toma una decisión respecto a si puede aprobarse o no para que lo utilicen los pacientes.
En los ensayos clínicos de fase 3 de terapia génica para la hemofilia se inscriben entre 50 y 150 personas con hemofilia y se realizan en puntos de estudio alrededor del mundo.
Fase 4:
La fase 4, algunas veces llamada ensayos poscomercialización, se realiza después de que un nuevo tratamiento ha recibido la aprobación reglamentaria y está disponible para los pacientes. Estos estudios permiten a los investigadores recolectar información adicional sobre los riesgos a más largo plazo (entre ellos efectos secundarios poco comunes) y los beneficios del tratamiento, así como su uso ideal en situaciones de la ‘vida real’.
Para obtener más información consulte el documento en formato PDF Cómo se prueban nuevas terapias en ensayos clínicos.

Cada ensayo clínico tiene un plan integral y detallado para realizar el estudio, conocido como el protocolo. El protocolo del ensayo clínico se elabora para responder preguntas de investigación específicas y para proteger la salud de los participantes en el estudio. Una agencia reguladora revisa y aprueba el protocolo antes de que pueda iniciarse el ensayo clínico. El protocolo de un ensayo clínico comprende la siguiente información:
El programa de desarrollo clínico de una nueva terapia o intervención determina si la nueva terapia es eficaz y segura. En el protocolo de un ensayo clínico, el criterio de valoración primario es la medida de resultados planeada que se considera más importante para evaluar la intervención o tratamiento. Dependiendo de la fase en la que se encuentre el ensayo clínico, el criterio de valoración primario podría enfocarse en la seguridad, tales como eventos adversos relacionados con el tratamiento y/o cambios en los valores iniciales en evaluaciones de laboratorio clínico; o en la eficacia, tales como tasa anualizada de hemorragias o niveles de factor.
Ejemplos de criterios de valoración de la seguridad y de la eficacia en ensayos clínicos para la hemofilia (incluso ensayos de terapia génica):
| Criterios de valoración de la seguridad | Criterios de valoración de la eficacia |
|---|---|
|
|
Es fundamental comprender los beneficios y los riesgos posibles al participar en un ensayo clínico.
| Posibles beneficios | Posibles riesgos |
|---|---|
| Podría tener acceso a un nuevo tratamiento antes de que esté disponible y podría ser de las primeras personas en recibir sus beneficios. | Podría experimentar efectos secundarios no deseados causados por el nuevo tratamiento. |
| Tendrá el apoyo de un equipo médico experto en hemofilia que monitoreará su salud estrechamente. | La nueva terapia pudiera no funcionar para usted o usted podría recibir un placebo, si se tratara de un estudio controlado con placebo. |
| Tendrá la oportunidad de desempeñar un papel activo en su salud y mejorar su propio tratamiento de la hemofilia. | La nueva terapia en estudio pudiera no ser mejor que su actual norma terapéutica. |
| Podría ayudar a personas diagnosticadas con hemofilia en el futuro, al contribuir al desarrollo de una posible terapia. | El ensayo pudiera tomarle más tiempo que el que le toma su tratamiento habitual; pudiera tener que hacerse más pruebas y más visitas al médico. |
Los posibles participantes en un ensayo clínico necesitan recordar que el propósito del ensayo clínico es estudiar un nuevo tratamiento o intervención.
A los pacientes se les invita a participar en ensayos clínicos a través de su equipo médico, con base en los criterios de elegibilidad del estudio. La elegibilidad se refiere a los requisitos clave que deben satisfacer las personas para poder participar en un ensayo clínico. Estos criterios ayudan a garantizar la seguridad de los participantes y a asegurarse de que las preguntas de investigación específicas que estudia el ensayo clínico se respondan de manera exacta.
Cada estudio tiene criterios de inclusión y criterios de exclusión.
En el caso de ensayos clínicos para la hemofilia, la gravedad y el tipo de hemofilia, la edad, la situación de inhibidores, y el historial profiláctico son factores comunes que determinan la elegibilidad. Cada ensayo clínico es único y los criterios de elegibilidad varían de un estudio al otro. Muchos ensayos clínicos se inician primero en adultos, antes de estudiar el nuevo tratamiento en niños. Por este motivo es muy común que la edad sea considerada un criterio de inclusión.
¿Desea obtener más información sobre criterios de elegibilidad?
Las PCH interesadas en participar en un ensayo clínico pasarán por un “proceso de selección” durante el cual el equipo de investigación determinará si la persona cumple con los criterios de elegibilidad del estudio. EL proceso de selección abarcará una revisión del historial médico de la persona y su estado médico actual, así como conversaciones sobre el papel y las responsabilidades de los participantes, y los posibles riesgos y beneficios de su participación.
Es importante comprender que no todas las personas interesadas en participar en un ensayo clínico tendrán la oportunidad de participar en el mismo. Esto puede suceder debido a que algún aspecto del historial médico de la persona no cumpla con los criterios de inclusión para el ensayo (por ej., el estudio podría estar reclutando solamente a PCH de cierta edad). Asimismo, cada ensayo clínico determinará el número de participantes que pueden inscribirse al mismo y dejará de aceptar participantes cuando haya alcanzado el número de participantes determinado en el estudio.
Repase un ejemplo (ficticio) de criterios de elegibilidad de ensayos clínicos para la hemofilia: Ejemplo 1 – Terapia génica para la hemofilia
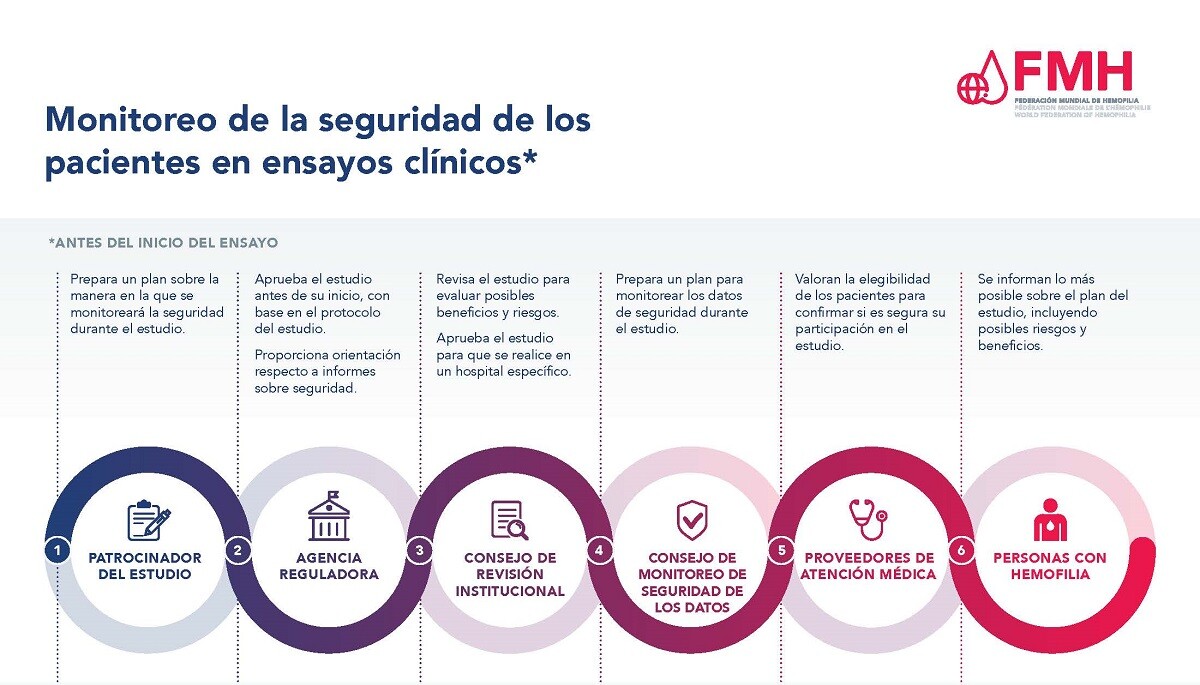
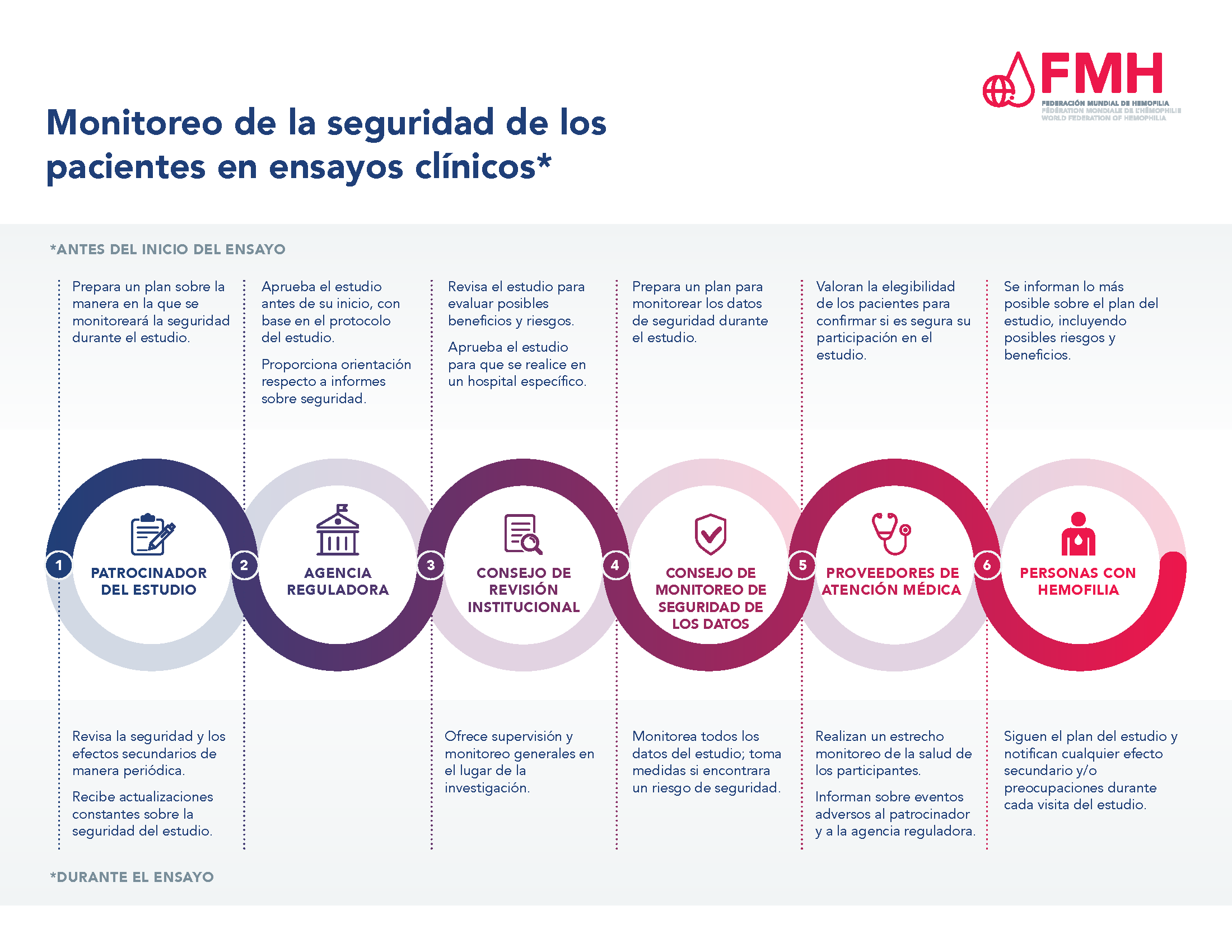
Garantizar la seguridad de los pacientes es de suma importancia durante el proceso del ensayo clínico. Hay muchos niveles de aprobación y monitoreo del estudio con el propósito de proteger la seguridad de los participantes en ensayos clínicos, y un sistema en pie para recolectar e informar resultados de seguridad durante un estudio.
Sin importar la fase del ensayo clínico, una agencia reguladora debe revisar y aprobar cada estudio antes de empezar a inscribir pacientes. El patrocinador del estudio, con frecuencia el fabricante del tratamiento bajo estudio, elabora un protocolo del estudio que incluye una sección sobre la forma en la que se monitoreará la seguridad del tratamiento a lo largo del estudio. Enseguida, este protocolo debe ser aprobado por una agencia reguladora, como la Administración de Alimentos y Medicamentos de Estados Unidos (FDA, por su sigla en inglés) o la Agencia Europea de Medicamentos (EMA, por su sigla en inglés). Una vez aprobado el estudio e identificados los sitios en los que se llevará a cabo, los sitios participantes deben presentar una solicitud y recibir la aprobación del protocolo del estudio del Consejo de Revisión Institucional que corresponda al hospital o centro de tratamiento de hemofilia en donde se realizará el estudio.
Una vez aprobado el estudio, un consejo de monitoreo de la seguridad de los datos, el patrocinador o la empresa fabricante del tratamiento en estudio, y los profesionales de la salud que participan en el mismo se encargan de monitorear la seguridad de los pacientes participantes. Las PCH inscritas en un estudio también desempeñan un papel en este aspecto y necesitan informar al equipo del estudio de su centro de tratamiento si llegaran a experimentar cualesquiera efectos secundarios o eventos de seguridad.
Para obtener más información consulte el documento PDF Monitoreo de la seguridad de los pacientes en ensayos clínicos.
Toda persona que participa voluntariamente en un ensayo clínico debe firmar un consentimiento informado antes de inscribirse en el estudio (en el caso de participantes menores de edad, su padre, madre o tutor pueden firmar un formulario de asentimiento). Un consentimiento informado proporciona a los posibles participantes información sobre el ensayo y explica los posibles riesgos y beneficios relacionados con su participación, antes de que decidan si desean o no participar en el estudio. El proceso para obtener el consentimiento informado abarca una conversación con el equipo del estudio antes de firmar el formulario.
En general, el proceso del consentimiento informado abarca lo siguiente:
La siguiente es una lista detallada de una agencia reguladora, en este caso de la FDA de EE. UU. Como parte del proceso de consentimiento informado, la siguiente información debe proporcionarse a cada posible sujeto de investigación antes de su inscripción a un ensayo clínico:
Para las personas que estén considerando participar en un ensayo clínico es sumamente importante contar con la mayor información posible sobre el mismo, sobre los riesgos y beneficios para los participantes, sus responsabilidades, la atención que pueden esperar y sobre el equipo que realizará el estudio. Es de esperarse que los posibles participantes tengan preguntas y es indispensable que obtengan respuestas a sus preguntas. Antes de reunirse con el equipo de investigación de un ensayo clínico podría ser útil escribir una lista de las preguntas que desea hacer.
Para obtener más información lea el documento PDF Preguntas que hacer antes de participar en un ensayo clínico.

Los miembros de equipo de atención médica desempeñan un papel fundamental a fin de garantizar que los posibles participantes en un estudio comprendan cabalmente lo que significa formar parte de un ensayo clínico.
| Componentes del proceso de consentimiento informado | Satisfacer las necesidades de posibles participantes en un ensayo clínico |
|---|---|
|
|
|
|
|
|
|
|
|
|
Las personas que están considerando participar en un ensayo clínico podrían tener preguntas sobre privacidad y la información de su atención médica. La confidencialidad se refiere a preservar la privacidad de los datos relacionados con la salud que permiten la identificación individual de personas que participan en ensayos clínicos. Esto abarca registros de investigación relacionados con identificadores (tales como el nombre), diagnóstico, pronóstico, tratamiento o cualquier otra información que pudiera relacionarse con un participante. En situaciones en las que los resultados de un ensayo clínico se divulgan en publicaciones revisadas por expertos, se ‘desidentifica’ la información relacionada con el paciente y se preserva la privacidad en dichos informes.
En el proceso de los ensayos clínicos participan muchos grupos diferentes de personas, y todos ellos tienen distintas responsabilidades y papeles. Los participantes en los ensayos clínicos y los proveedores de atención médica tienen diferentes papeles a lo largo del proceso del ensayo clínico. Es importante que ambos estén formados y comprendan plenamente sus papeles.
Consulte las papelas y responsabilidades de cada uno de estos grupos a continuación.
Para obtener más información consulte el documento PDF Papel del paciente y del proveedor de atención médica durante ensayos clínicos.

Cada país tiene su propia autoridad reguladora, con sus propios reglamentos o leyes, para realizar ensayos clínicos. La autoridad reguladora revisa y aprueba los protocolos de ensayos clínicos antes del inicio de los estudios y se asegura de que todos los ensayos clínicos se apeguen a los reglamentos nacionales. La agencia reguladora interactúa con los investigadores a lo largo de todo el proceso del ensayo clínico y en última instancia revisa todos los datos sobre seguridad y eficacia de un programa de desarrollo clínico a fin de determinar si el nuevo tratamiento/intervención debiera aprobarse y ponerse a disposición del público.
Persona, empresa, institución, grupo u organización que asume la responsabilidad de iniciar, gestionar y/o financiar un ensayo clínico.
Grupo de científicos, médicos, no científicos y defensores de pacientes que revisa y aprueba el plan detallado para un ensayo clínico. El objetivo de los CRI es proteger a las personas que participan en un ensayo clínico. En países fuera de EE. UU., este grupo es llamado comité de ética.
Grupo de científicos independientes que monitorea la seguridad e integridad de un ensayo clínico.
La(s) persona(s) a cargo de un ensayo clínico. El investigador principal o ‘IP’ a menudo es un médico.
Persona responsable de realizar ensayos clínicos, de acuerdo con los principios de buenas prácticas de laboratorio y bajo la orientación de un investigador principal.
Miembros del equipo de atención médica que participan en la ejecución de un ensayo clínico. Entre ellos, médicos, asistentes médicos, personal de enfermería, farmacéuticos, científicos y otras personas que apoyan a los participantes en el ensayo a lo largo del proceso del estudio, realizan pruebas, evaluaciones/valoraciones durante las visitas del estudio, recolectan datos, e implementan todos los aspectos del protocolo del ensayo clínico.
Personas que satisfacen los criterios de elegibilidad y participan en un ensayo clínico.