Les études cliniques font généralement appel à des personnes volontaires, à des participants ou à des échantillons (sang ou autres tissus) prélevés sur des êtres humains, dans le but de faire progresser les connaissances médicales. Il existe deux grands types d’études cliniques : les essais cliniques (également appelés études interventionnelles) et les études observationnelles.
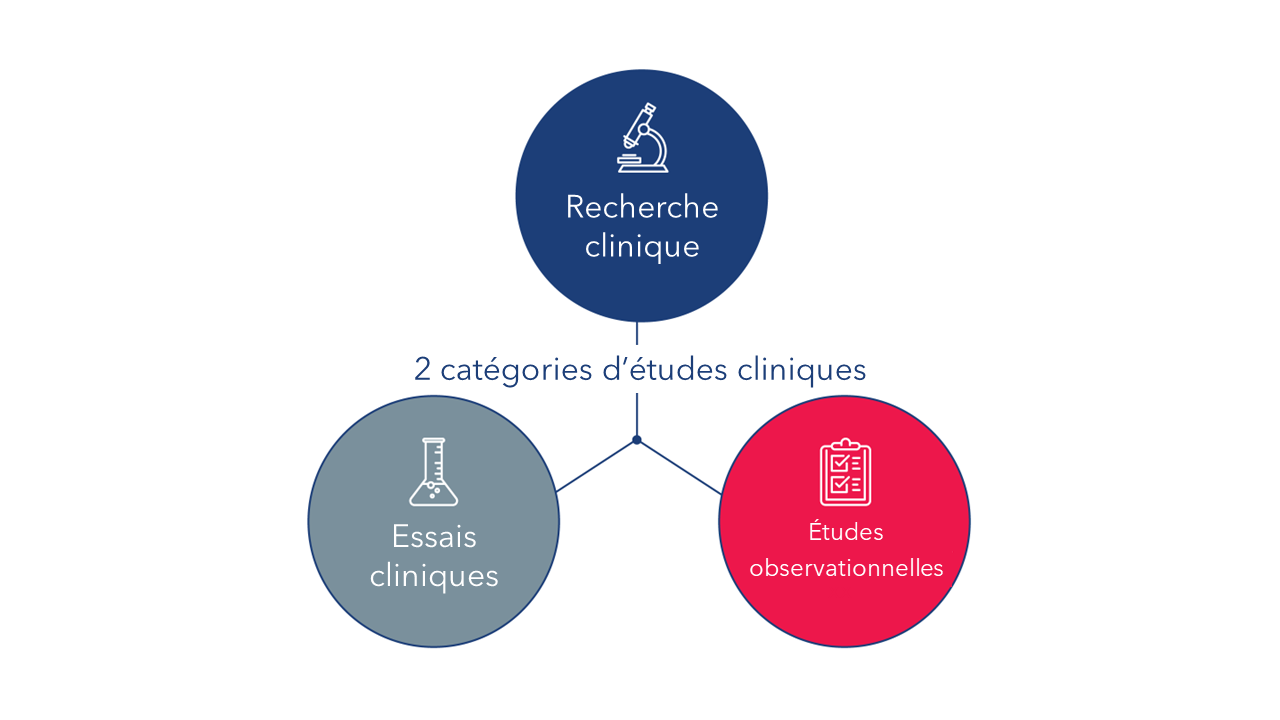
Un essai clinique est un type d’étude dans lequel les chercheurs testent de nouvelles méthodes de prévention, de détection ou de traitement des maladies. Les participants à un essai clinique font l’objet d’interventions spécifiques conformément à un protocole détaillé pour l’essai clinique en question. De telles interventions peuvent prendre la forme de produits médicaux, comme des médicaments ou des dispositifs, des procédures chirurgicales ou des changements de comportement, comme le régime alimentaire d’un participant.
Les essais cliniques peuvent comparer une nouvelle méthode de traitement à une méthode standard déjà disponible, à un placebo ou à une approche sans intervention. Les essais cliniques sont la principale méthode utilisée par les chercheurs pour déterminer si un nouveau traitement est sûr et efficace. Pour qu’un médicament puisse être prescrit par les médecins, il doit d’abord être testé dans une série d’essais cliniques, appelés phase 1, phase 2 et phase 3. Après les essais cliniques de phase 3, les données des essais cliniques sont soumises à un organisme de réglementation qui détermine alors si l’utilisation de ce médicament peut être approuvée.
Il existe différents types d’essais cliniques :
Les termes suivants sont souvent utilisés dans le cadre d’essais cliniques :
Une étude observationnelle est un type d’étude dans lequel les chercheurs observent les effets d’une intervention sur un groupe de participants, sans intervenir. Contrairement à un essai clinique, les participants ne font pas l’objet d’interventions spécifiques par l’investigateur, mais peuvent recevoir un traitement qui fait déjà partie de leurs protocoles de soins habituels. Les chercheurs peuvent alors évaluer les associations entre les interventions et les résultats de santé, chez les personnes qui les prennent dans le cadre d’un protocole de soins standard. De tels résultats peuvent conduire à une étude plus approfondie dans le cadre d’un essai clinique. Il existe plusieurs types d’études observationnelles. Un registre de patients est un type d’étude observationnelle.
Un registre de patients est un système organisé qui utilise des méthodes d’étude observationnelle pour collecter des données sur les traitements, les résultats cliniques et le bien- être d’une population définie par une maladie, une condition ou une exposition particulière.
Dans le domaine de l’hémophilie, il existe deux registres de patients importants :
1. Registre mondial des troubles de la coagulation
Le Registre mondial des troubles de la coagulation (RMTC) est un système de saisie de données en ligne qui fournit une plateforme à des centres de traitement de l’hémophilie du monde entier visant à recueillir des données uniformes et normalisées sur les patients et à orienter la pratique clinique. Avec le consentement éclairé du patient, le RMTC stocke des données pseudonymisées sur la maladie de l’individu, comme le type et la sévérité de l’hémophilie, les symptômes, le traitement et les résultats en matière de santé.
Les professionnels de la santé qui participent au RMTC peuvent l’utiliser pour suivre et surveiller les progrès de leurs patients et orienter leurs soins cliniques. Ces données pseudonymisées et confidentielles peuvent également être utilisées pour aider les chercheurs à répondre à des questions importantes sur les disparités dans l’offre de soins dans le monde et contribuer à faire avancer les initiatives en matière de plaidoyer et de politique de santé.
Cliquer ici pour en savoir plus sur le RMTC.
2. Registre de la thérapie génique de la Fédération mondiale de l’hémophilie
Dans le cadre d’une collaboration internationale, la FMH a établi un registre mondial des patients traités par thérapie génique, le registre de la thérapie génique (RTG) de la FMH . L’objectif du RTG est de fournir une base de données solide et scientifiquement valide, accessible à tous les professionnels de santé qui prennent en charge des personnes atteintes d’hémophilie recevant une thérapie génique, partout dans le monde. Les données recueillies par le RTG de la FMH seront utilisées pour évaluer la sécurité et l’efficacité à long terme de la thérapie génique chez les personnes atteintes d’hémophilie.
Cliquer ici pour en savoir plus sur le RTG de la FMH.
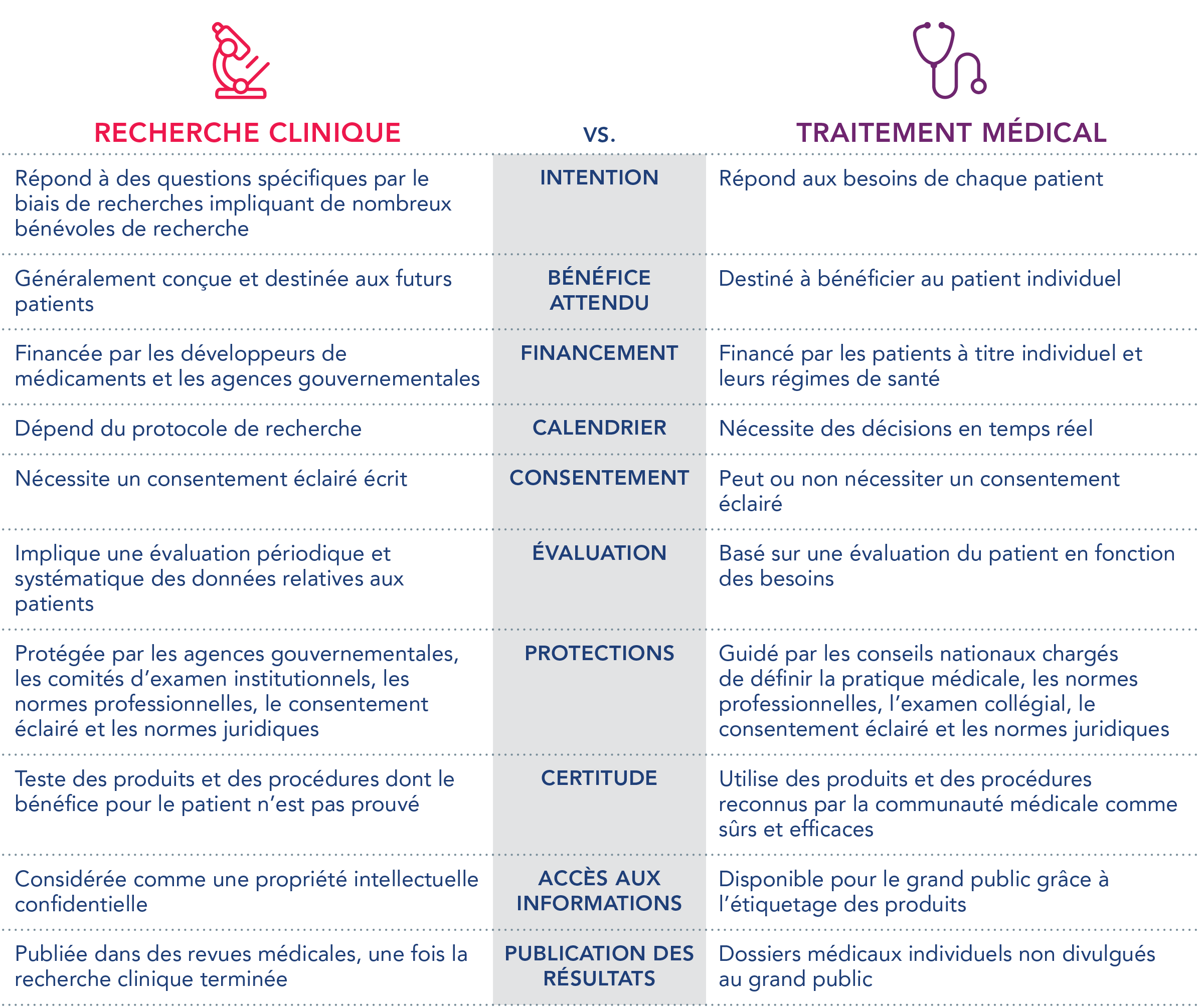
Le tableau suivant peut vous aider à comprendre en quoi la recherche clinique est différente d’un traitement médical.
Pour qu’un médicament puisse être prescrit par les médecins, il doit d’abord être testé au cours d’une série de phases d’essais cliniques, puis évalué et approuvé par un organisme de réglementation. Chacune des phases d’un essai clinique a un objectif bien spécifique et suit une chronologie de la phase 1 à la phase 4.
Phase 1 :
C’est la première fois qu’un nouveau traitement expérimental est testé sur des personnes. L’objectif de la phase 1 est d’évaluer la sécurité et les effets secondaires associés au traitement. Cette phase est souvent menée sur un petit groupe de volontaires sains. Cependant, dans le cas de maladies rares, comme l’hémophilie, la phase 1 fait appel à des personnes atteintes d’hémophilie. Cette phase dure généralement plusieurs mois et environ 70 % des traitements testés passent à la phase suivante. Le développement clinique ne va pas au delà de la phase 1 pour les 30 % restants, les résultats n’ayant pas été satisfaisants s’agissant de la sécurité.
Pour la thérapie génique dans le domaine de l’hémophilie, la phase 1 fait intervenir entre 10 et 30 personnes atteintes d’hémophilie.
Phase 2 :
L’objectif de la phase 2 est de continuer à évaluer la sécurité et les effets secondaires associés au traitement, mais aussi de tester l’efficacité et de déterminer les dosages les plus efficaces. La phase 2 est généralement menée sur un plus grand nombre de participants que lors de la phase 1 et inclut des personnes atteintes de la maladie. Dans certaines situations, les 2 premières phases de l’essai clinique sont combinées et qualifiées de phase 1/2. En règle générale, la phase 2 dure entre quelques mois et deux ans. On estime que 33 % des traitements testés lors de la phase 2 sont stoppés par manque d’efficacité ou problèmes de sécurité.
Pour la thérapie génique dans le domaine de l’hémophilie, la phase 2 d’un essai clinique fait intervenir entre 10 et 5 personnes atteintes d’hémophilie. Ce nombre est plus faible que pour d’autres maladies car l’hémophilie est une maladie rare. Pour les maladies rares comme l’hémophilie, les phase 1 et 2 sont souvent combinées. L’industrie pharmaceutique espère ainsi accélérer les délais de développement des nouveaux médicaments.
Phase 3 :
L’objectif de la phase 3 est de confirmer l’efficacité du traitement, de surveiller les effets secondaires et de comparer le nouveau traitement aux traitements standard ou similaires. La phase 3, parfois appelée « phase pivot », est menée avec un grand nombre de personnes atteintes de la maladie et dans de nombreux sites différents (nationaux et internationaux, selon l’étude). La phase 3 est parfois randomisée et souvent en double aveugle. Elle dure généralement de 12 à 48 mois et constitue la dernière étape avant la soumission d’une demande d’autorisation de mise sur le marché auprès d’une agence de régulation. On estime que 25 % des essais de phase 3 passent à la phase 4.
Les données de l’étude pivot de phase 3, et parfois les données des phases 1 et 2, sont ensuite soumises à un organisme de réglementation pour examen. Il effectue des analyses indépendantes sur la sécurité et l’efficacité du traitement et prend la décision d’approuver ou non l’utilisation du traitement par les patients.
Pour la thérapie génique dans le domaine de l’hémophilie, la phase 3 fait intervenir entre 50 et 150 personnes atteintes d’hémophilie et est conduite dans plusieurs sites d’étude dans le monde entier.
Phase 4 :
La phase 4, parfois appelée phase de pharmacovigilance, est menée une fois que le nouveau traitement a reçu l’approbation réglementaire et est disponible pour les patients. Ces études permettent aux chercheurs de recueillir des informations supplémentaires sur les risques à long terme (y compris les effets secondaires rares) et les avantages du traitement, ainsi que sur son utilisation optimale dans des situations « réelles ».
Pour plus d’informations, veuillez consulter le document PDF intitulé Comment les nouvelles thérapies sont testées dans le cadre d’essais cliniques ?

Chaque essai clinique comporte un plan détaillé et complet pour la réalisation de l’essai, appelé protocole. Le protocole de l’essai clinique est développé pour répondre à des questions de recherche spécifiques et pour protéger la santé des participants à l’étude. Avant le démarrage de tout essai clinique, le protocole est examiné et approuvé par une agence de régulation. Les informations contenues dans un protocole d’essai clinique comprennent :
Le programme de développement clinique d’une nouvelle thérapie ou intervention permet de déterminer si le nouveau traitement est efficace et sûr. Dans un protocole d’essai clinique, le principal critère d’évaluation primaire est la mesure des résultats prévus. Selon la phase de l’essai clinique, le principal critère d’évaluation peut être axé sur la sécurité, comme les événements indésirables liés au traitement et/ou les changements par rapport aux valeurs de départ dans les évaluations de laboratoire clinique, ou sur l’efficacité, comme le taux de saignement annualisé ou le taux de facteur.
Exemples de critères d’évaluation de la sécurité et de l’efficacité dans les essais cliniques en hémophilie (y compris les essais de thérapie génique) :
| Critères de sécurité | Critères d’efficacité |
|---|---|
|
|
Il est indispensable de comprendre les bénéfices et les risques potentiels qu’implique toute participation à un essai clinique.
| Bénéfices potentiels | Risques potentiels |
|---|---|
| Avoir accès à un nouveau traitement avant sa commercialisation et être parmi les premiers à en bénéficier | Effets indésirables possibles dûs au nouveau traitement |
| Bénéficier du soutien d’une équipe médicale spécialisée en hémophilie assurant un suivi étroit de votre état de santé | Nouveau traitement inefficace ou administration d’un placébo dans le cas d’une étude contrôlée par placébo |
| Jouer un rôle actif dans la prise en charge de sa propre santé et améliorer la prise en charge de son hémophilie | Aucun bénéfice particulier du nouveau traitement par rapport au traitement standard |
| Aider les personnes diagnostiquées dans le futur en contribuant au développement d’un traitement potentiel de l’hémophilie | Essai plus chronophage que le traitement habituel avec davantage d’examens et de consultations |
Les participants potentiels à un essai clinique doivent se rappeler que le but de l’essai clinique est d’étudier un nouveau traitement ou une nouvelle intervention.
En fonction des critères d’éligibilité de l’étude, c’est l’équipe soignante qui invite le patient à participer à un essai clinique. Par éligibilité, on entend les principales exigences à satisfaire pour que les personnes puissent participer à un essai clinique. Ces critères permettent de garantir la sécurité des participants et de s’assurer que les questions de recherche spécifiques étudiées dans l’essai clinique puissent trouver une réponse précise.
Chaque étude comporte des critères d’inclusion et des critères d’exclusion.
Pour les essais cliniques dans le domaine de l’hémophilie, la sévérité et le type d’hémophilie, l’âge, la présence ou non d’inhibiteurs et les antécédents en matière de prophylaxie sont des critères habituels permettant de déterminer l’éligibilité. Chaque essai clinique est unique et les critères d’éligibilité varient d’une étude à l’autre. De nombreux essais cliniques commencent chez les adultes, avant d’étudier le nouveau traitement chez les enfants. C’est pourquoi il est très fréquent de voir l’âge figurer parmi les critères d’inclusion.
Vous voulez en savoir plus sur les critères d’éligibilité ?
Les personnes atteintes d’hémophilie souhaitant participer à un essai clinique seront soumises à un « processus de sélection » au cours duquel l’équipe de recherche déterminera si l’individu remplit les critères d’éligibilité de l’étude. Le processus de sélection comprendra un examen des antécédents médicaux et de l’état de santé actuel de la personne atteinte d’hémophilie, ainsi que des discussions sur les rôles et responsabilités des participants, et sur les risques et bénéfices potentiels d’une éventuelle participation.
Il est important de comprendre que toutes les personnes intéressées par un essai clinique n’auront pas la possibilité d’y participer, par exemple, parce que ses antécédents médicaux ne répondent pas aux critères d’inclusion de l’essai (une étude peut ainsi ne recruter que des individus d’un certain âge). En outre, chaque essai clinique aura un nombre spécifique de participants à recruter ; une fois ce nombre obtenu, il cessera d’accepter de nouveaux participants à l’étude.
Découvrez un exemple (fictif) de critères d’éligibilité pour des essais cliniques dans le domaine de l’hémophilie : Exemple 1 de thérapie génique en hémophilie
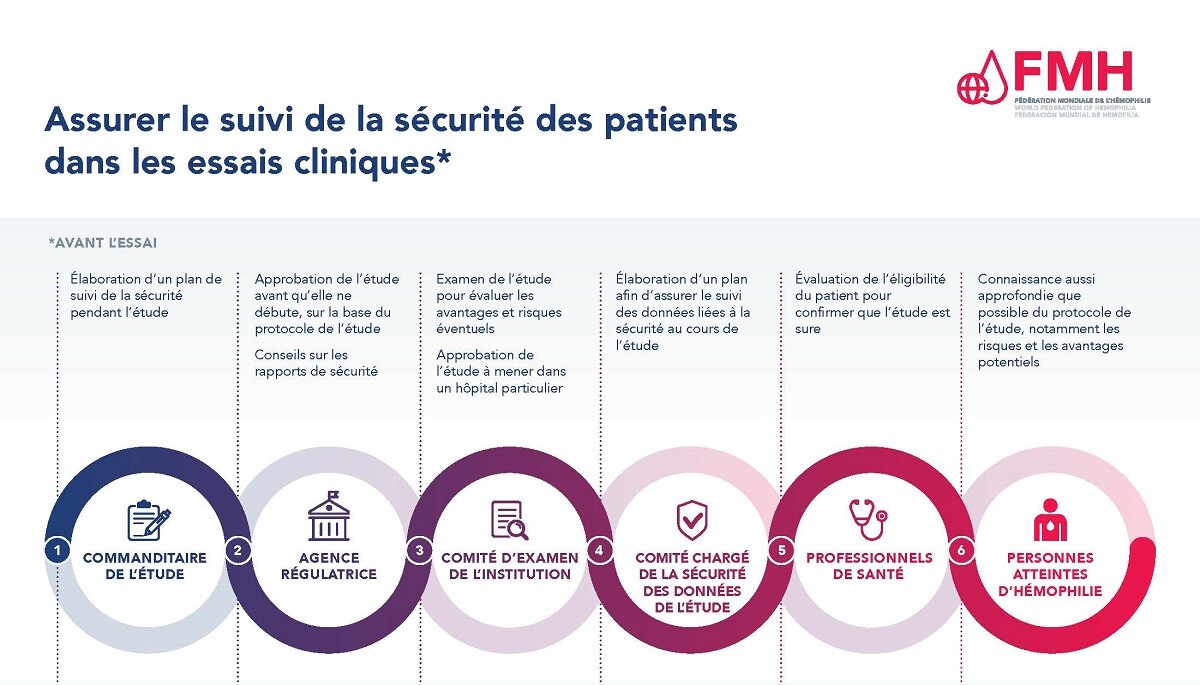
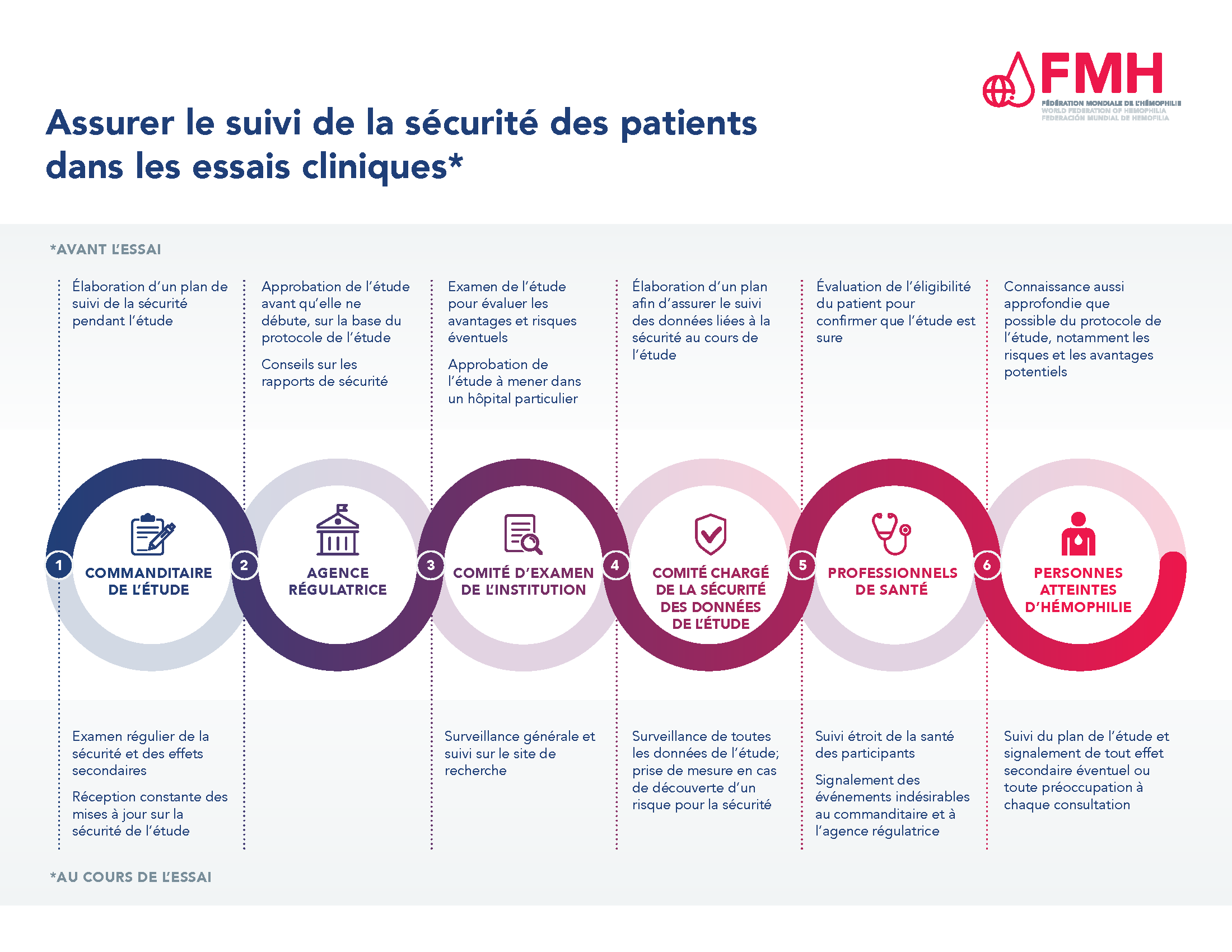
Assurer la sécurité des patients est d’une importance capitale au cours du processus d’essai clinique. Il existe de nombreux niveaux d’approbation et de contrôle des études qui visent à protéger la sécurité des participants dans le cadre des essais cliniques, ainsi qu’un système de collecte et de communication des résultats en matière de sécurité au cours d’une étude.
Quelle que soit la phase d’un essai clinique, chaque étude doit être examinée et approuvée par une agence de régulation avant tout recrutement de patients. Le promoteur de l’étude, souvent le fabricant du traitement à l’étude, élabore un protocole d’étude qui comprend une section sur la manière dont la sécurité du traitement sera contrôlée tout au long de l’étude. Le protocole de l’étude doit ensuite être approuvé par une agence de régulation, tel que la Food and Drug Administration (FDA) ou l’Agence européenne des médicaments (EMA). Une fois l’étude approuvée et les sites d’étude identifiés, les sites participants doivent soumettre et faire approuver le protocole de l’étude par le conseil d’examen institutionnel utilisé par l’hôpital ou le centre de traitement de l’hémophilie où l’étude sera menée.
Une fois l’étude approuvée, la sécurité des patients participants fait l’objet d’un suivi tout au long de l’étude par un comité de surveillance de la sécurité des données, par le promoteur ou la société qui fabrique le traitement à l’étude, et par les professionnels de santé impliqués. Les personnes atteintes d’hémophilie recrutées pour une étude ont également un rôle à jouer. Elles doivent informer l’équipe de l’étude dans leur centre de traitement si elles ressentent des effets secondaires ou des événements liés à la sécurité.
Pour plus d’informations, veuillez consulter le document PDF Assurer le suivi de la sécurité des patients dans les essais cliniques.
Chaque participant se portant volontaire pour participer à un essai clinique doit signer un formulaire de consentement éclairé avant tout recrutement (pour les enfants mineurs, un parent doit signer). Un consentement éclairé fournit aux participants potentiels des informations sur l’essai clinique et présente les risques et les avantages potentiels associés à la participation avant qu’une personne ne décide de participer. Le processus d’obtention du consentement éclairé implique une discussion avec l’équipe de l’étude avant la signature du formulaire.
En règle générale, le processus de consentement éclairé inclut les étapes suivantes :
La liste suivante est celle fournie par une agence de régulation, ici la FDA américaine. Dans le cadre du processus de consentement éclairé, ces informations doivent être fournies à chaque participant potentiel avant tout recrutement à un essai clinique :
Pour les personnes qui envisagent de participer à un essai clinique, il est extrêmement important d’en apprendre le plus possible sur l’essai clinique, les soins escomptés, le coût et l’équipe qui conduira l’essai. Il est tout à fait normal d’avoir des questions et il est essentiel d’obtenir des réponses. Il peut être utile de rédiger une liste de questions à poser avant de rencontrer l’équipe de recherche chargée de l’essai clinique.
Pour en savoir plus, veuillez consulter le document PDF intitulé Questions à poser avant de participer à un essai clinique.

Les membres de l’équipe soignante ont un rôle essentiel à jouer pour s’assurer que les participants potentiels à l’étude comprennent parfaitement ce qu’implique leur participation à un essai clinique.
| Composantes du processus du consentement éclairé | Répondre aux besoins des participants potentiels à un essai clinique |
|---|---|
|
|
|
|
|
|
|
|
|
|
Toute personne envisageant de participer à un essai clinique peut tout naturellement s’interroger sur la confidentialité des informations qui concerne son état de santé. Par confidentialité, on entend la protection du caractère privé des informations relatives à la santé permettant d’identifier les personnes qui participent à un essai clinique. Cela comprend les ressources liées à l’identité (comme le nom), au diagnostic, au pronostic, au traitement ou à toute autre information en lien avec un participant. Lorsque les résultats d’un essai clinique sont publiés dans une revue évaluée collégialement, les informations relatives aux patients sont anonymisées et la confidentialité préservée.
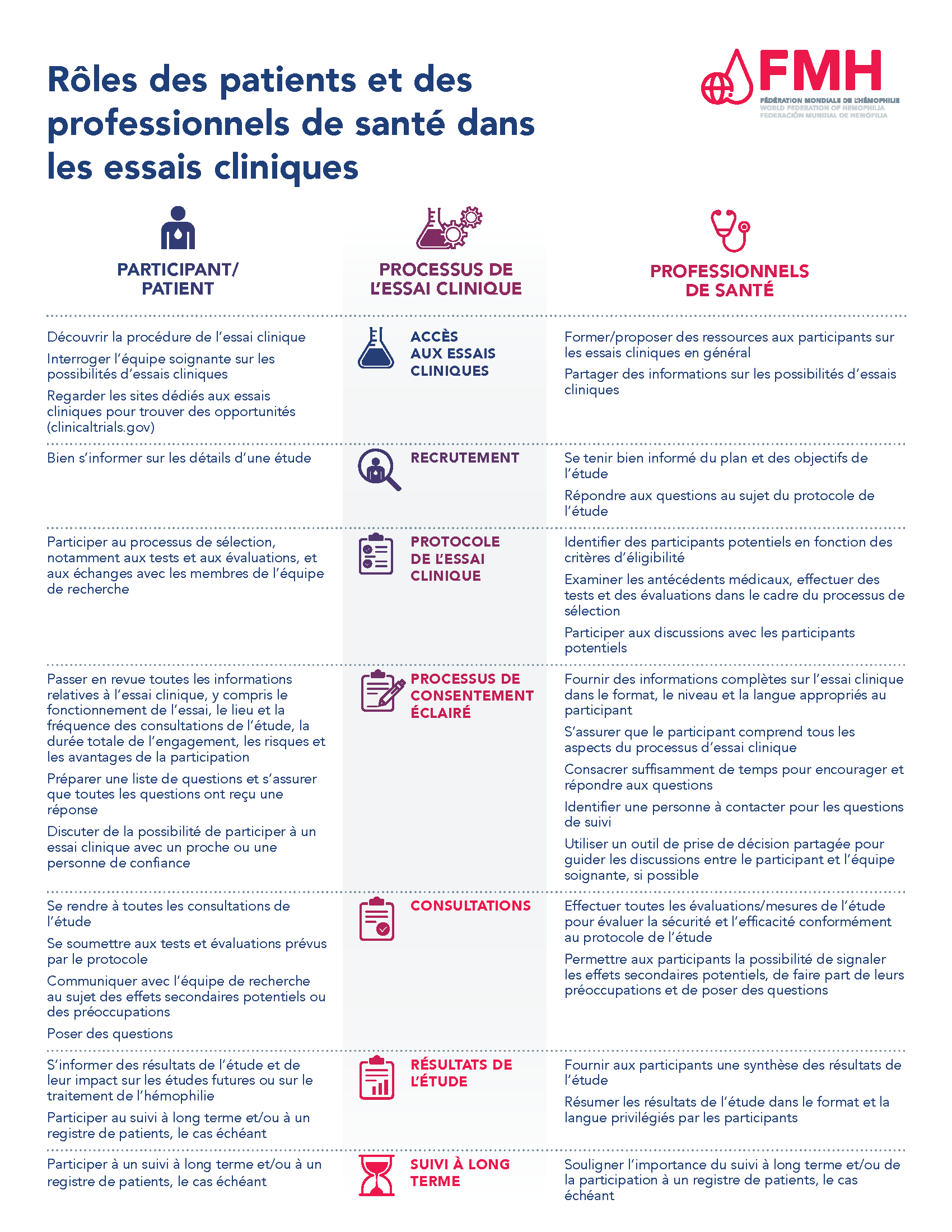
De nombreux groupes de personnes sont impliqués dans le processus d’essai clinique, et ils ont tous des responsabilités et des rôles différents. Les participants aux essais cliniques et les professionnels de santé ont des rôles différents tout au long du processus d’essai clinique. Il est important que les uns et les autres soient informés et comprennent parfaitement leurs rôles.
Veuillez consulter les rôles et responsabilités de chacun de ces groupes ci-dessous.
Pour plus d’informations, veuillez consulter le document PDF intitulé Rôles des patients et des professionnels de santé dans les essais cliniques.
Chaque pays dispose de sa propre agence de régulation avec ses propres réglementations ou lois régissant la conduite des essais cliniques. L’agence de régulation examine et approuve les protocoles d’essais cliniques avant le début des études et s’assure que tous les essais cliniques respectent la réglementation nationale. Il échange avec les chercheurs tout au long du processus d’essai clinique et examine en dernier ressort toutes les données relatives à la sécurité et à l’efficacité d’un programme de développement clinique afin de déterminer si le nouveau traitement peut obtenir une autorisation de mise sur le marché.
Personne, entreprise, institution, groupe ou organisation responsable du lancement, de la gestion et/ou du financement d’un essai clinique.
Groupe de scientifiques, de médecins, de non-scientifiques et de défenseurs des patients qui examine et approuve le plan détaillé d’un essai clinique. Le Comité d’examen institutionnel a pour but de protéger les personnes qui participent à un essai clinique. Dans les pays autres que les États-Unis, ce groupe est appelé Comité d’éthique.
Groupe de scientifiques indépendants qui surveillent la sécurité et l’intégrité d’un essai clinique.
Personne en charge d’un essai clinique. L’investigateur principal est souvent un médecin.
Personne chargée de mener des essais cliniques conformément aux principes des bonnes pratiques de laboratoire et sous la direction de l’investigateur principal.
Membres de l’équipe soignante participant à la réalisation d’un essai clinique. Il s’agit de médecins, d’infirmiers, d’assistants médicaux, de pharmaciens, de scientifiques et d’autres personnes qui soutiennent les participants tout au long du processus, effectuent des tests, des évaluations/bilans lors des consultations, collectent des données et mettent en œuvre tous les aspects du protocole de l’essai clinique.
Individus qui répondent aux critères d’éligibilité et participent à un essai clinique.