Clinical studies typically involve human volunteers, participants, or samples (blood or other tissues) from humans with the intent of advancing medical knowledge. There are two main types of clinical studies: clinical trials (also called interventional studies) and observational studies.
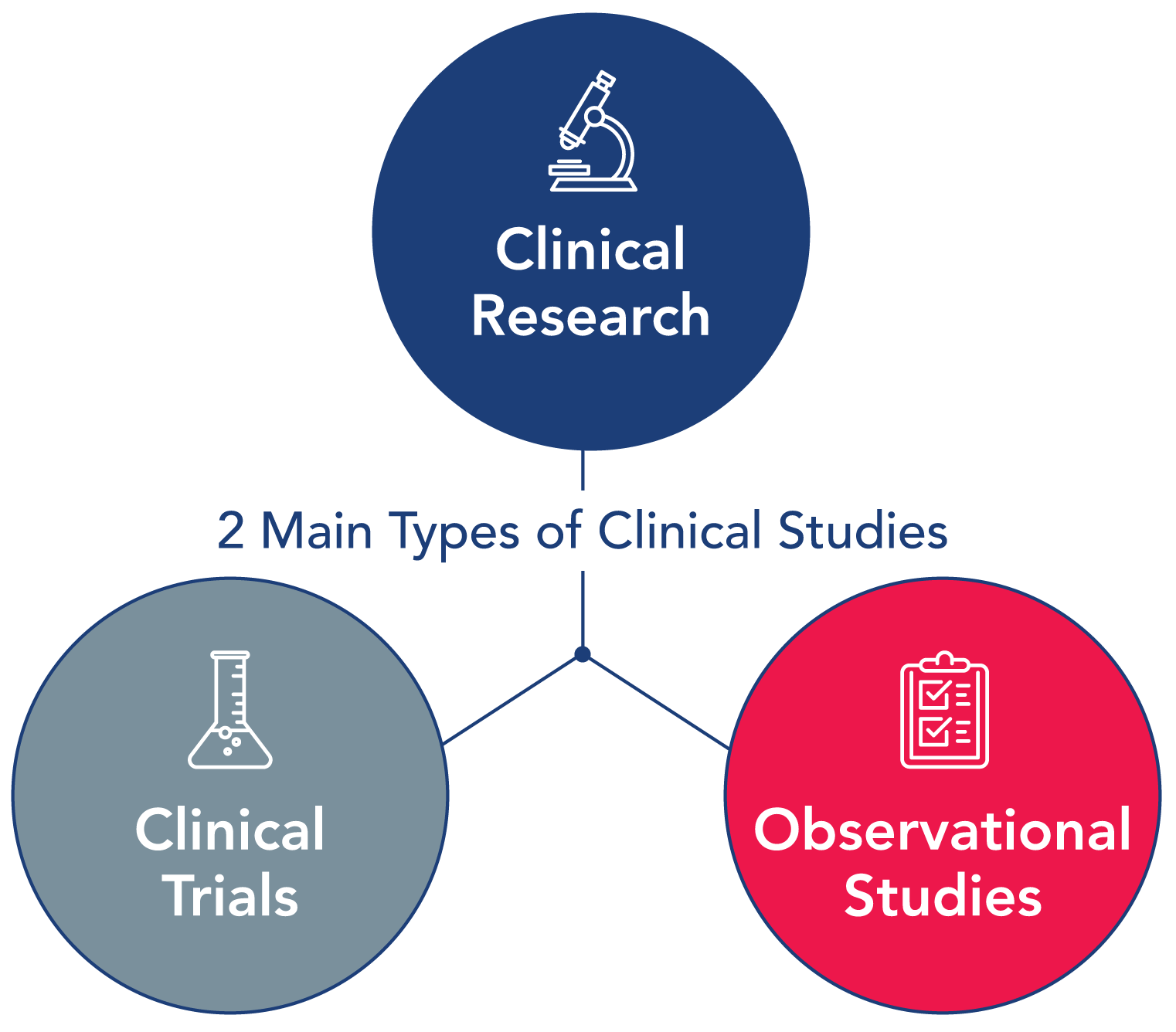
A clinical trial is a type of research study in which researchers test new ways to prevent, detect, or treat diseases. Participants in a clinical trial receive specific interventions according to a detailed protocol for that clinical trial. Such interventions may be medical products, such as drugs or devices, surgical procedures, or changes to behavior, such as a participant’s diet.
Clinical trials may compare a new treatment method to a standard one that is already available, to a placebo, or to no intervention. Clinical trials are the main method that researchers use to find out if a new treatment is safe and efficacious. For a drug to become a medicine that doctors can prescribe, it must first be tested in a series of clinical trials, known as Phase 1, Phase 2, and Phase 3. Following Phase 3 clinical trials, data from the clinical trials is submitted to a regulatory agency who then determines if this drug should be approved for use.
There are different types of clinical trials:
The following terms are often used in describing clinical trials:
An observational study, is a type of research study in which researchers observe the effects of an intervention in a group of participants, without intervening. Participants are not assigned to specific interventions by the investigator, as in a clinical trial, but may be receiving a treatment that is already part of their routine medical care. Researchers can then assess associations between interventions and health outcomes, among people taking it as part of standard care. Such findings may lead to further investigation in a clinical trial. There are several different types of observational studies. A patient registry is one type of observational study.
A patient registry is an organized system that uses observational study methods to collect data on treatments, clinical outcomes, and the well-being of a population defined by a particular disease, condition, or exposure.
For the hemophilia community, two important patient registries are:
1. World Bleeding Disorders Registry
The World Bleeding Disorders Registry (WBDR) is a web-based data entry system that provides a platform for a network of hemophilia treatment centers around the world to collect uniform and standardized patient data, and to guide clinical practice. With informed consent from the patient, the WBDR stores de-identified data about the person’s disease, such as hemophilia type and severity, symptoms, treatment and health outcomes.
Health care professionals participating in the WBDR can use the WBDR to track and monitor their patient’s progress and guide their clinical care. These de-identified and confidential data can also be used to help researchers answer important questions about disparities in care globally and help advance advocacy/health policy initiatives.
2. World Federation of Hemophilia Gene Therapy Registry
Through an international collaborative effort, the WFH has developed a global gene therapy patient registry, the WFH Gene Therapy Registry (GTR). The aim of the GTR is to provide a robust, scientifically valid database, available to all healthcare providers treating people with hemophilia who receive gene therapy, anywhere in the world. The data collected through the WFH GTR will be used to assess the long-term safety and efficacy of gene therapy in persons with hemophilia.
The following table can be helpful to see how clinical research is different from a medical treatment.
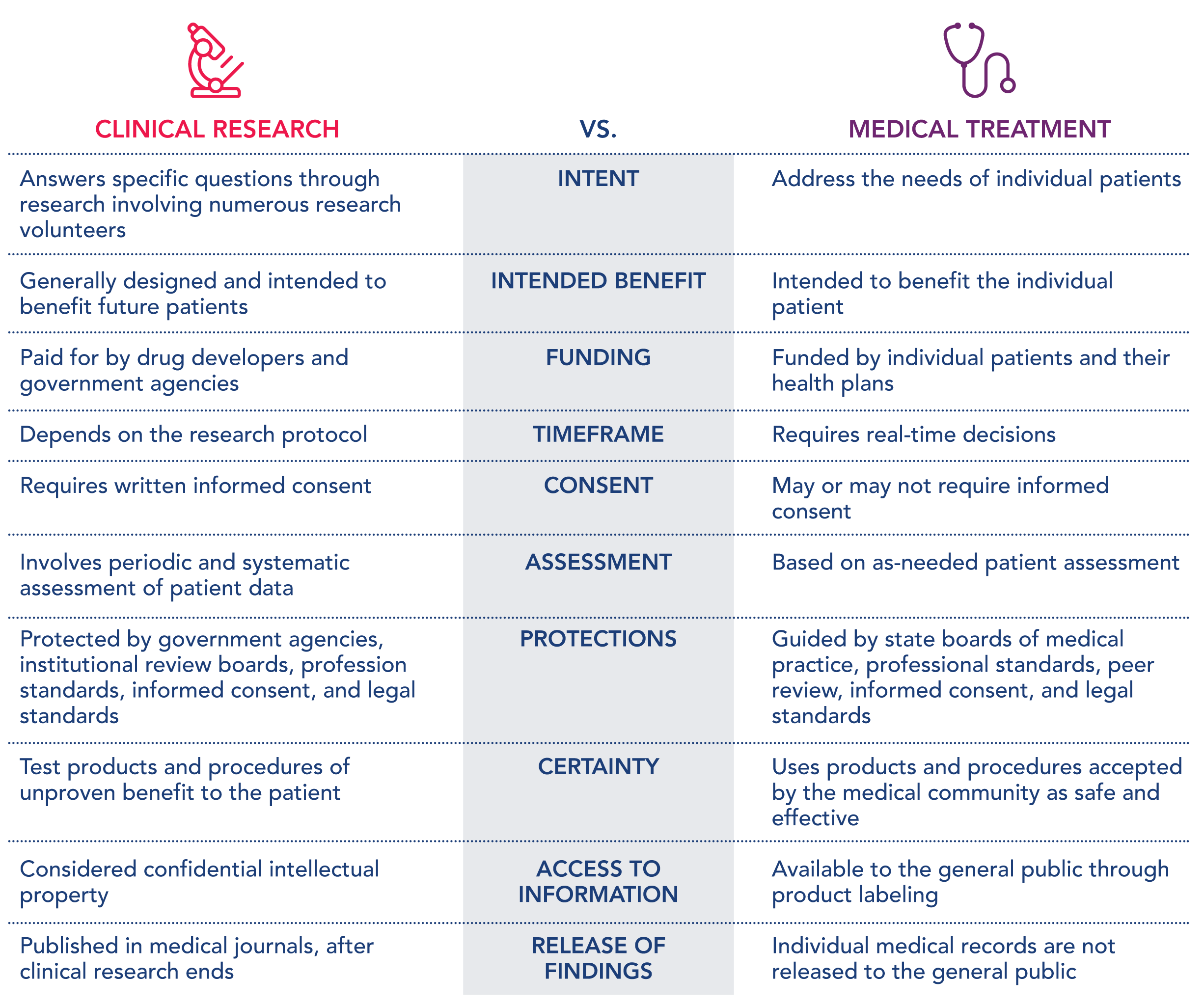
For a drug to become a treatment that doctors can prescribe, it must first be tested in a series of clinical trial phases, and then assessed and approved by a regulatory agency. Each clinical trial phase has a purpose, and the phases progress in order from Phase 1 to Phase 4.
Phase 1:
This is the first time a new investigational treatment is tested in people. The purpose of Phase 1 clinical trials is to assess the safety and side effects associated with the treatment. They are often conducted in a small group of healthy volunteers. However, in rare diseases, such as hemophilia, Phase I trials are usually conducted in people with hemophilia. Phase 1 clinical trials are typically several months long and approximately 70% of therapies tested in Phase 1 trials move to the next phase. Clinical development does not proceed beyond Phase 1 for the other 30% of therapies that are not shown to be safe in Phase 1.
Phase 1 hemophilia gene therapy clinical trials enroll between 10-30 persons with hemophilia.
Phase 2:
The purpose of Phase 2 clinical trials is to continue to test the safety and side effects associated with the treatment, but also test the efficacy and determine the most effective dosages. Phase 2 trials are usually conducted in more participants than a Phase 1 trial, and include people who have the disease. In some situations, Phase 1 and Phase 2 trials are combined and referred to as Phase 1/2. Phase 2 clinical trials are typically several months to 2 years long. It is estimated that 30% of therapies tested in Phase 2 trials move to the next phase. Clinical development does not proceed beyond Phase 2 if the treatment is not shown to be efficacious or there are safety concerns.
Phase 2 hemophilia gene therapy clinical trials enroll between 10-50 persons with hemophilia. This number is smaller than in other disease states because hemophilia is a rare disease. Phase I and 2 clinical trials are often combined for rare diseases such as hemophilia. By combining trial phases, drug manufacturers hope to speed up the development timelines for new drugs.
Phase 3:
The purpose of Phase 3 clinical trials is to confirm efficacy of the treatment, monitor side effects, and compare the new treatment with standard or similar treatments. Phase 3 trials, sometimes called a ‘pivotal trial,’ are conducted on a large number of people with the disease and in many different study locations (national and international, depending on the study). Phase 3 clinical trials are sometimes randomized and often double-blinded. Phase 3 clinical trials are typically 1 to 4 years long and are the last step before submitting an application to a regulatory agency for approval. An estimated 25% of Phase 3 trials move to Phase 4 studies.
Data from the Phase 3 pivotal study, and sometimes data from Phase I and 2 clinical trials, are submitted to a regulatory agency for review. The regulatory agency conducts independent analyses on the safety and efficacy of the treatment and makes a decision on whether a treatment is approved for use by patients or not.
Phase 3 hemophilia gene therapy clinical trials enroll between 50-150 persons with hemophilia and are conducted in study locations around the world.
Phase 4:
Phase 4, sometimes called post-marketing trials, are conducted after a new treatment has received regulatory approval and is available to patients. These studies allow researchers to gather additional information about the longer-term risks (including rare side effects) and benefits of the treatment, as well as optimal use in ‘real-world’ situations.
Please see the How New Therapies Get Tested in Clinical Trials PDF for more information.

Each clinical trial has a detailed, comprehensive plan for conducting the trial called the protocol. The clinical trial protocol is developed to answer specific research questions and to protect the health of the study participants. The protocol is reviewed and approved by a regulatory agency before a clinical trial can begin. Information contained in a clinical trial protocol includes:
The clinical development program of a new therapy or intervention determines if the new therapy is effective and safe. In a clinical trial protocol, the primary endpoint is the planned outcome measure that is the most important for evaluating the intervention or treatment. Depending on the phase of the clinical trial, the primary endpoint may be focused on safety, such as treatment-related adverse events and/or changes from baseline in clinical laboratory evaluations; or efficacy, such as annualized bleeding rate or factor level.
Examples of safety and efficacy endpoints in hemophilia clinical trials (including gene therapy trials):
| Safety Endpoints | Efficacy Endpoints |
|---|---|
|
|
It is critical to understand the potential benefits and risks of participating in a clinical trial.
| Potential Benefits | Potential Risks |
|---|---|
| You may have access to a new treatment before it is available and could be among the first to benefit | You may experience unwanted side effects of the new treatment |
| You will have the support of an expert hemophilia care team who will monitor your health closely | The new therapy may not work for you or you may receive placebo if it is a placebo-controlled study |
| You will have an opportunity to play an active role in your health and improve your own hemophilia management | The new therapy being studied may not be better than your current standard of care |
| You may help future people diagnosed with hemophilia by contributing to the development of a potential therapy | The trial could take more of your time than your usual treatment; you may have more doctor visits and tests |
Potential clinical trial participants need to remember that the purpose of the clinical trial is to study a new treatment or intervention.
Patients are invited to join clinical trials through their health care team, based on the eligibility criteria of the study. Eligibility refers to the key requirements that must be met for people to participate in a clinical trial. These criteria help to ensure the safety of participants and ensure that the specific research questions being studied in the clinical trial may be answered accurately.
Each study has both inclusion criteria and exclusion criteria.
For hemophilia clinical trials, severity and type of hemophilia, age, inhibitor status, and history of prophylaxis are common factors determining eligibility. Each clinical trial is unique and eligibility criteria vary from study to study. Many clinical trials start in adults first, before studying the new treatment in children. For this reason, it is very common to see age listed as an inclusion criterion.
Want to know more about eligibility criteria?
People with hemophilia who are interested in participating in a clinical trial will go through a “screening process” where the research team will determine if that person meets the eligibility criteria of the study. The screening process will include a review of the PWH’s medical history and current medical status, as well as discussions on the roles and responsibilities of participants, and the potential risks and benefits of participating.
It is important to understand that not everyone who has an interest in a clinical trial will have the opportunity to participate. This can happen because some aspect of a person’s medical history does not meet the inclusion criteria for the trial (for example, a study may only be enrolling PWH of a certain age). Further, each clinical trial will have an estimated number of participants for planned enrollment, when a clinical trial has the required number of people enrolled they will stop accepting additional study participants.
Review a (fictional) example of eligibility criteria for hemophilia clinical trials: Example 1 Gene Therapy in Hemophilia
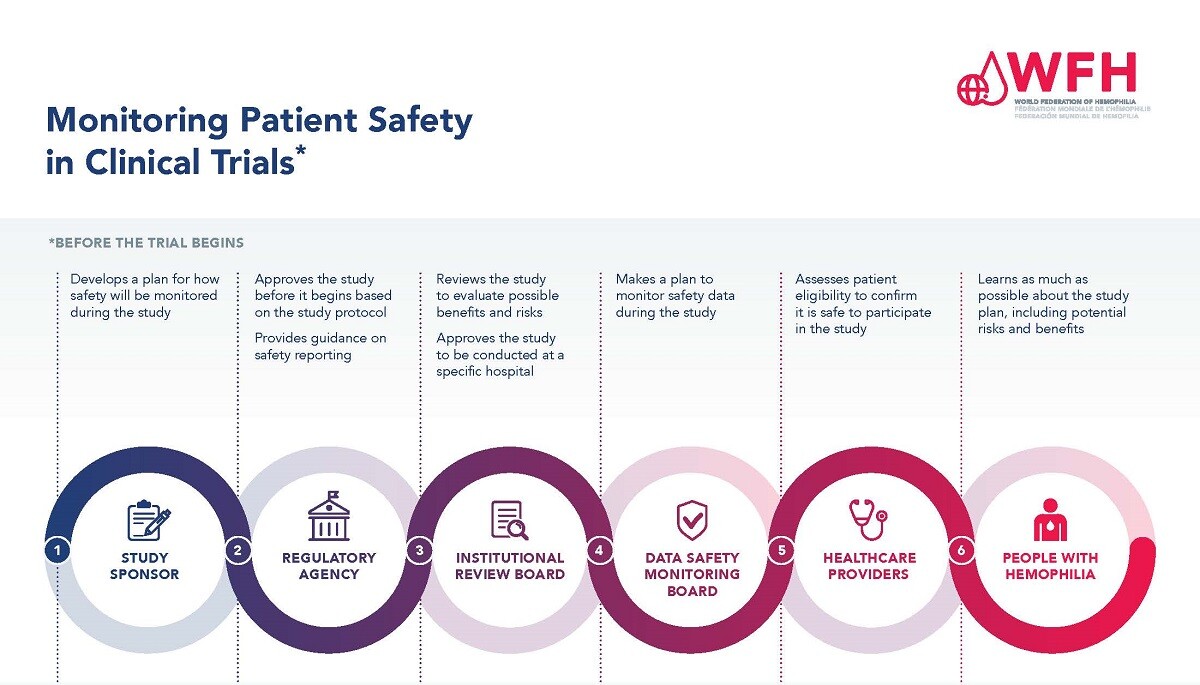
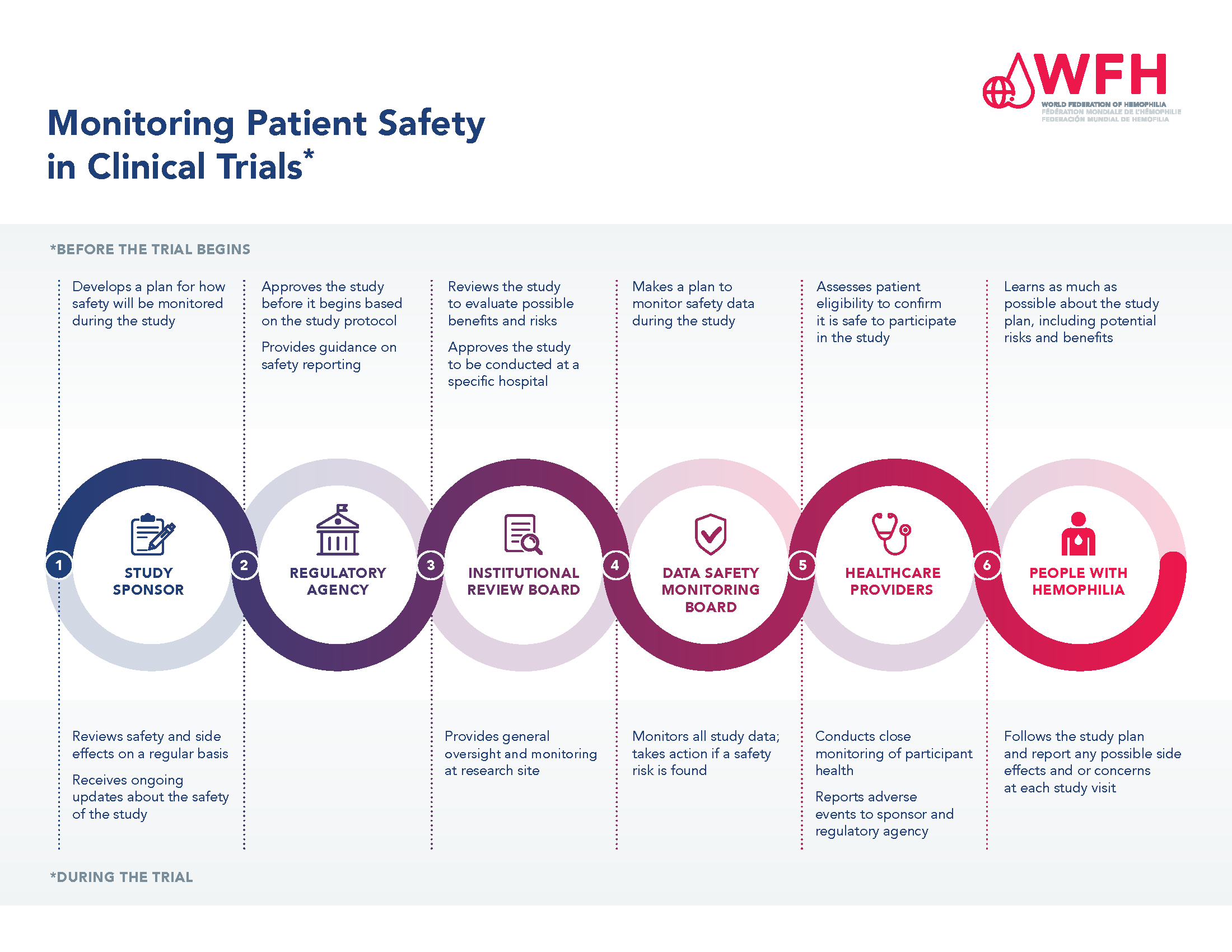
Ensuring patient safety is of paramount importance during the clinical trial process. There are many levels of study approval and monitoring to protect the safety of participants in clinical trials, and a system in place for collecting and reporting safety outcomes during a study.
Regardless of the phase of a clinical trial, each study must be reviewed and approved by a regulatory agency before patients can be enrolled. The study sponsor, often the manufacturer of the treatment under study, develops a study protocol that includes a section on how the safety of the treatment will be monitored throughout the study. The study protocol must then be approved by a regulatory agency, such as the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA). Once a study is approved and study sites are identified, participating sites must submit and receive approval of the study protocol to the Institutional Review Board used by the hospital or hemophilia treatment center where it will be carried out.
Once a study is approved, the safety of patients participating is monitored throughout the study by a Data Safety Monitoring Board, the sponsor or the company manufacturing the study treatment, and by the healthcare professionals involved. The PWH enrolled in a study also have a role to play. PWH participating in trials need to inform the study team at their treatment center if they experience any side effects or safety events.
Please see the Monitoring Patient Safety in Clinical Trials PDF for more information.
Every participant who volunteers to participate in a clinical trial must sign an informed consent form prior to being enrolled in the study (or a parent can sign an assent form for their child in the case of minor participants). An informed consent provides potential participants information about the trial and explains the potential risks and benefits associated with participating before a person decides to participate. The process of obtaining informed consent involves a discussion with the study team prior to signing the form:
In general, the informed consent process involves the following:
The following is a detailed list from a regulatory agency, in this example the U.S. FDA. As part of the informed consent process, this information must be given to each potential research subject before enrolling in a clinical trial:
For people considering participation in a clinical trial, it is extremely important to learn as much as possible about the clinical trial, the risks and benefits to the participants, the responsibilities of participants, the care expected, and the team that will be conducting the trial. It is expected that you will have questions, and essential that your questions are answered. It may be helpful to write down a list of questions to ask before meeting with a clinical trial research team.
Please see the Questions to ask before participating in clinical trial PDF for more information.

Members of the healthcare team have a vital role in ensuring that potential study participants fully understand what it will mean to be part of a clinical trial.
| Components of the Informed Consent Process | Meeting the Needs of Potential Clinical Trial Participants |
|---|---|
|
|
|
|
|
|
|
|
|
|
Individuals who are considering participation in a clinical trial may have questions about their health care information and privacy. Confidentiality refers to maintaining privacy with individually identifiable health-related information for persons who participate in clinical trials. This includes research records related to identification (such as name), diagnosis, prognosis, treatment, or any other information that could be linked to a participant. In situations in which results of a clinical trial are published in a peer-reviewed journal, patient related information is de-identified, and privacy is maintained in such reporting.
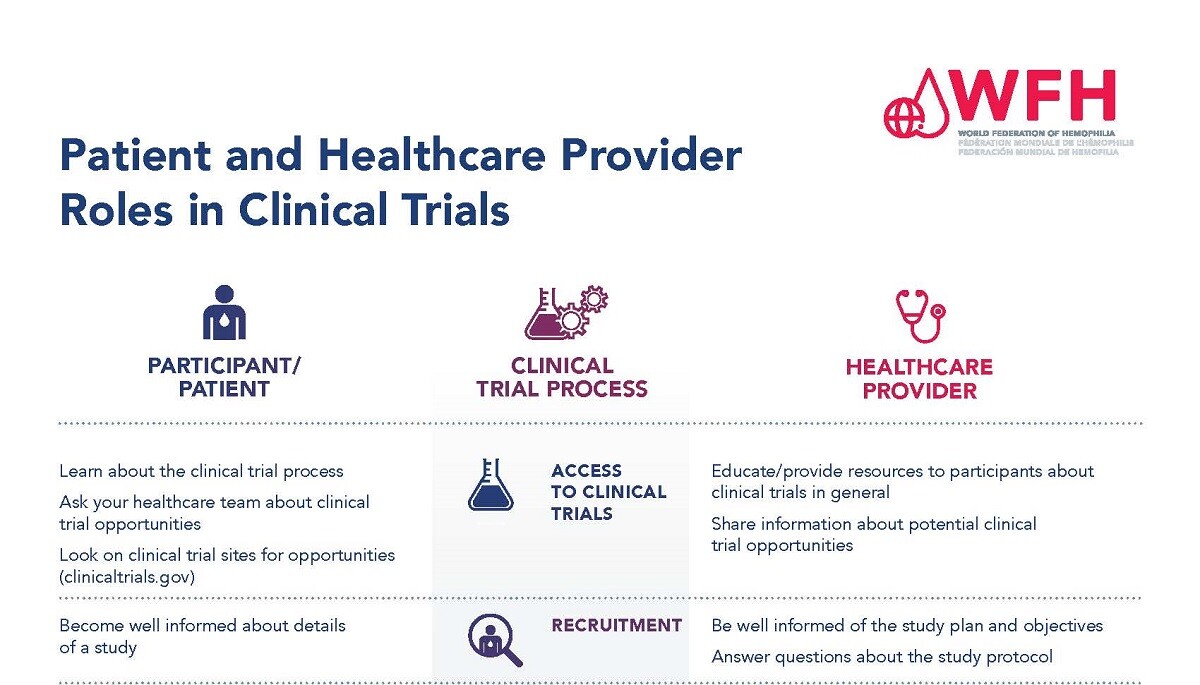
Many different groups of people are involved in the clinical trial process, and they all have different responsibilities and roles. Participants and health care providers play an important role throughout the duration of the clinical trial process. It is important that both participants and health care providers fully understand their roles.
Please see the roles and responsibilities of each of these groups below.
Please see the Patient and Healthcare Provider Roles in Clinical Trials PDF for more information.

Each country has its own regulatory authority with its own regulations, or laws, for conducting clinical trials. The regulatory authority reviews and approves clinical trial protocols before studies begin and ensures that all clinical trials follow national regulations. The regulatory agency interacts with researchers throughout the clinical trial process, and ultimately reviews all of the safety and efficacy data from a clinical development program in order to determine if the new treatment/intervention should be approved and publicly available.
A person, company, institution, group, or organization that takes responsibility for initiation, management, and/or financing of a clinical trial.
A group of scientists, doctors, non-scientists, and patient advocates that reviews and approves the detailed plan for a clinical trial. IRBs are meant to protect the people who take part in a clinical trial. In countries outside of the United States, this group is called an Ethics Committee.
A group of independent scientists who monitor the safety and integrity of a clinical trial.
The person(s) in charge of a clinical trial. The principal investigator, or ‘PI,’ is often a medical doctor.
A person responsible for conducting clinical trials according to principles of good laboratory practice (GLP) and under the guidance of a principal investigator.
Members of the health care team involved in conducting a clinical trial. This includes medical doctors, nurses, physician assistants, pharmacists, scientists, and others who support trial participants throughout the clinical trial process, perform tests, evaluations/assessments at study visits, collect data, and carry out all aspects of the clinical trial protocol.
Individuals who meet the eligibility criteria and participate in a clinical trial.