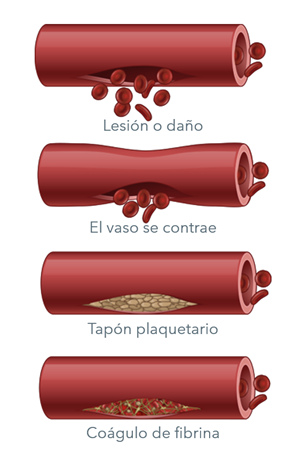
Los factores de la coagulación son proteínas de la sangre que controlan las hemorragias. Cuando un vaso sanguíneo se lesiona, sus paredes se contraen para limitar el flujo de sangre al área afectada. Entonces, pequeñas células sanguíneas llamadas plaquetas se pegan al sitio de la lesión y se distribuyen a lo largo de la superficie del vaso sanguíneo para detener la hemorragia. Al mismo tiempo, pequeños sacos al interior de las plaquetas liberan señales químicas que atraen a otras plaquetas al sitio de la lesión y provocan su aglutinamiento para formar lo que se conoce como tapón plaquetario.
En la superficie de estas plaquetas activadas, muchos factores de la coagulación diferentes trabajan en conjunto en una serie de complejas reacciones químicas (conocidas como la cascada de la coagulación) a fin de formar un coágulo de fibrina. El coágulo funciona como una red que detiene la hemorragia.
Los factores de la coagulación circulan en la sangre sin estar activados. Cuando un vaso sanguíneo sufre una lesión se inicia la cascada de la coagulación y cada factor de la coagulación se activa en un orden específico para dar lugar a la formación del coágulo sanguíneo. Los factores de la coagulación se identifican con números romanos (por ejemplo, factor VIII o FVIII). La forma activada del factor de coagulación se escribe agregando la letra “a” (por ejemplo, factor VIIIa).
La cascada de la coagulación constituye una serie de vías que convergen en el factor X, un factor de la coagulación indispensable para la liberación de fibrina y la formación del coágulo.
En personas con una deficiencia de factor de coagulación hay un factor de coagulación (o, muy raramente, más de uno) que falta, se encuentra presente en concentraciones muy bajas, o no funciona adecuadamente. Esto afecta el proceso normal de coagulación de la sangre, lo cual dificulta la formación de un coágulo.
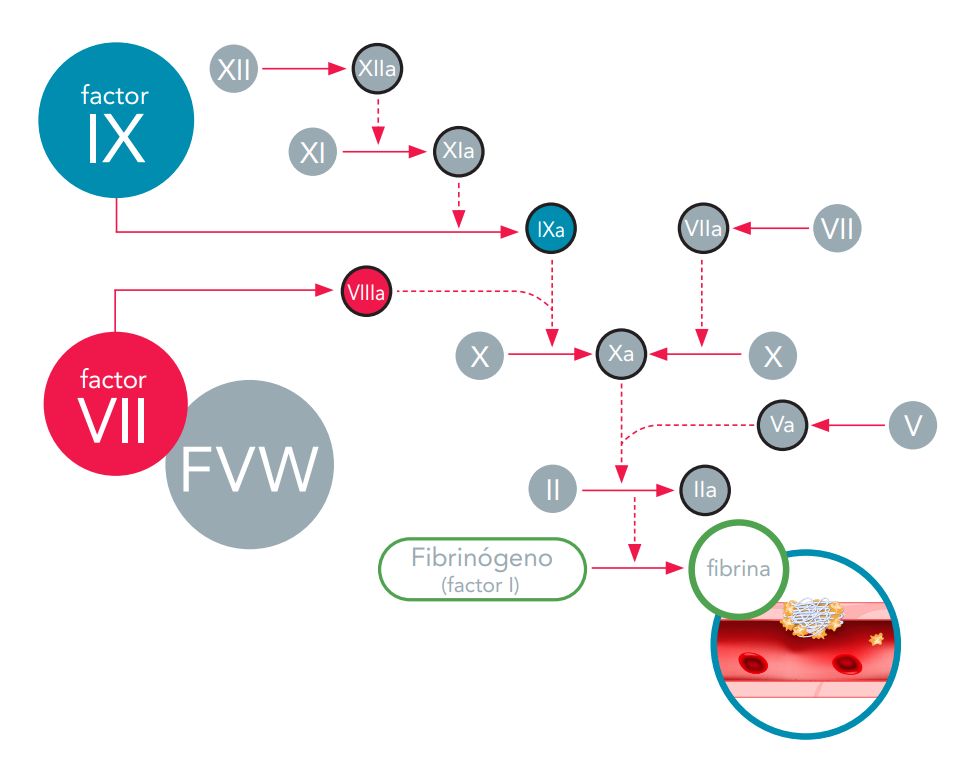
Si cualquiera de los factores de la coagulación faltara o no funcionara adecuadamente, la cascada de la coagulación se interrumpe. Cuando esto ocurre, no se forma el coágulo sanguíneo y la hemorragia continua durante más tiempo del que debiera.
Las deficiencias de factor VIII y factor IX son conocidas como hemofilia A y B, respectivamente. La deficiencia del factor Von Willebrand (FVW) o sus defectos se conocen como enfermedad de Von Willebrand (EVW). Las deficiencias poco comunes de factores de la coagulación son trastornos hemorrágicos en los que uno o más de los otros factores (es decir, los factores I, II, V, V+VIII, VII, X, XI o XIII) faltan o no funcionan adecuadamente. El conocimiento que se tiene de estos trastornos es menor porque se diagnostican muy raramente. Todas las deficiencias de factor son enfermedades poco comunes, y las deficiencias poco comunes de factores de la coagulación se consideran enfermedades sumamente poco comunes porque afectan a un número todavía menor de personas.
Los seres humanos tienen veintitrés pares de cromosomas: veintidós pares de cromosomas autosómicos (también llamados autosomas) y un par de cromosomas sexuales (X o Y). A diferencia de la hemofilia, que se debe a mutaciones en el cromosoma X, las deficiencias poco comunes de factores de la coagulación se deben a mutaciones en los autosomas.
La deficiencia de factor I (fibrinógeno) es un trastorno de la coagulación hereditario causado por un problema con el factor I. Debido a que el cuerpo produce menos fibrinógeno del que debiera o debido a que el fibrinógeno no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma. La deficiencia de factor I es un término amplio que abarca diversos trastornos relacionados, conocidos como defectos congénitos del fibrinógeno.
La afibrinogenemia es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos.
La hipofibrinogenemia, la disfibrinogenemia y la hipodisfibrinogenemia pueden ser ya sea recesivas (ambos padres son portadores del gene) o dominantes (solo uno de los padres es portador y transmisor del gene).
Todos los tipos de deficiencia de factor I afectan tanto a hombres como a mujeres.
Los síntomas de la deficiencia de factor I varían dependiendo del tipo de trastorno que tenga cada persona.
Debido a que el fibrinógeno desempeña un papel central en la coagulación, la ausencia total de fibrinógeno da lugar a defectos hemostáticos graves, desde hemorragias que ponen en peligro la vida hasta trombosis. Los episodios hemorrágicos son los síntomas más frecuentes; generalmente no son previsibles y pueden ser hemorragias graves que ponen en peligro la vida.
Las personas con afibrinogenemia podrían presentar hemorragias tales como las siguientes:
*La incidencia de eventos trombóticos es mayor en niños pequeños.
Los síntomas son similares a los observados en la afibrinogenemia. En general, entre menor cantidad de factor I tenga una persona en la sangre, mayor será la frecuencia y/o la gravedad de los síntomas.
Los síntomas dependen de la manera en la que funcione el fibrinógeno (que se encuentra presente en concentraciones normales). Algunas personas no presentan ningún síntoma. Otras personas tienen hemorragias (como las observadas en casos de afibrinogenemia), y otras presentan señales de trombosis (coágulos sanguíneos anormales en los vasos sanguíneos) en lugar de hemorragias.
Los síntomas son variables y dependen de la cantidad de fibrinógeno que se produzca y de cómo funcione.
La deficiencia de factor I se diagnostica mediante diversas pruebas sanguíneas, entre ellas una prueba específica que mide la cantidad de fibrinógeno en la sangre. No obstante, bajas concentraciones de fibrinógeno o su función anormal podrían ser señales de otras enfermedades, tales como trastornos hepáticos o renales, los cuales deberían descartarse antes de diagnosticarse un trastorno de la coagulación. Las pruebas diagnósticas debe realizarlas un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación.
Hay tres tratamientos disponibles para la deficiencia de factor I. Todos se fabrican a partir de plasma humano.
También puede administrarse tratamiento para evitar la formación de coágulos sanguíneos, ya que esta complicación puede ocurrir después de la terapia de reemplazo con fibrinógeno.
Muchas personas con hipofibrinogenemia o disfibrinogenemia no necesitan tratamiento. Los periodos menstruales abundantes en mujeres con deficiencia de factor I pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia de factor II (protrombina) es un trastorno de la coagulación hereditario causado por un problema con el factor II. Debido a que el cuerpo produce menos protrombina de la que debiera o debido a que la protrombina no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor II es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia de factor II es muy poco común.
La deficiencia de factor II puede heredarse junto con otras deficiencias de factor (véase la sección sobre “Deficiencia combinada de factores de la coagulación dependientes de la vitamina K”). También puede presentarse en una etapa posterior de la vida, como resultado de enfermedad hepática, deficiencia de vitamina K o consumo de ciertos medicamentos como la warfarina, un fármaco anticoagulante. La deficiencia de factor II adquirida es más común que la deficiencia heredada.
Los síntomas de la deficiencia de factor II son diferentes en cada persona. Generalmente, entre menor sea la concentración de factor II en la sangre de una persona, mayor será la frecuencia y/o gravedad de los síntomas.
Las personas con deficiencia de factor II podrían presentar hemorragias tales como las siguientes:
La deficiencia de factor II se diagnostica mediante diversas pruebas sanguíneas. El médico necesitará medir el tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activada (TTPa). Si ambas pruebas señalaran un tiempo prolongado será necesario medir las concentraciones de factores I, II, V, VII, IX y X en la sangre. Las pruebas diagnósticas debe realizarlas un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación.
Hay dos tratamientos disponibles para la deficiencia de factor II. Ambos se fabrican a partir de plasma humano.
Los periodos menstruales abundantes en mujeres con deficiencia de factor II pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia de factor V es un trastorno de la coagulación hereditario causado por un problema con el factor V. Debido a que el cuerpo produce menos factor V del que debiera o debido a que el factor V no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor V es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia de factor V es muy poco común.
Los síntomas de la deficiencia de factor V son generalmente leves, y algunas personas podrían no presentar ningún síntoma. No obstante, los niños con una deficiencia grave de factor V podrían presentar hemorragias a una edad muy temprana. Algunas personas con deficiencia de factor V han presentado hemorragias en el sistema nervioso central (cerebro y médula espinal) en una fase muy temprana de la vida.
Las personas con deficiencia de factor V podrían presentar hemorragias tales como las siguientes:
La deficiencia de factor V se diagnostica mediante varias pruebas sanguíneas que debe realizar un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación. El médico necesitará medir el tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activada (TTPa). Si ambas pruebas señalaran un tiempo prolongado será necesario medir las concentraciones de factores I, II, V, VII, IX y X en la sangre. En las personas con concentraciones anormales de factor V también deberían verificarse las concentraciones de factor VIII a fin de descartar la posibilidad de una deficiencia combinada de factor V y factor VIII, lo cual constituye un trastorno totalmente diferente.
El tratamiento para la deficiencia de factor V generalmente solo se requiere en casos de hemorragias graves o antes de cirugías. El plasma fresco congelado (PFC) o el PFC con patógenos reducidos es el tratamiento habitual porque no existe un concentrado que contenga exclusivamente factor V. Las transfusiones de plaquetas, que contienen factor V, también constituyen una opción en algunos casos.
Los periodos menstruales abundantes en mujeres con deficiencia de factor V pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia combinada de factor V y factor VIII es un trastorno de la coagulación hereditario causado por bajas concentraciones de factor V y de factor VIII. Debido a que la cantidad de estos factores en el cuerpo es menor a la normal, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma. Esta deficiencia combinada constituye una deficiencia totalmente diferente de la deficiencia de factor V y de la deficiencia de factor VIII (hemofilia A).
La deficiencia combinada de factor V y factor VIII es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia es muy poco común y, en casos extremadamente raros, la deficiencia de factor VIII podría heredarse por separado solamente de uno de los padres.
En la mayoría de los casos, el trastorno es causado por un solo defecto genético que afecta la capacidad del cuerpo para transportar factor V y factor VIII fuera de la célula y hacia el torrente sanguíneo, y no por un problema con el gene de cualquiera de los factores.
La deficiencia combinada de factor V y factor VIII no parece provocar más hemorragias que las que se presentarían si solo uno u otro de los factores estuviera afectado. Los síntomas de la deficiencia combinada de factor V y factor VIII generalmente son leves.
Las personas con deficiencia combinada de factor V y factor VIII podrían presentar hemorragias tales como las siguientes:
La deficiencia combinada de factor V y factor VIII se diagnostica mediante diversas pruebas sanguíneas para determinar si las concentraciones de ambos factores son inferiores a las normales. Estas pruebas debería realizarlas un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación.
Hay tres tratamientos disponibles para la deficiencia combinada de factor V y factor VIII.
Los periodos menstruales abundantes en mujeres con deficiencia combinada de factor V y factor VIII pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia de factor VII es un trastorno de la coagulación hereditario causado por un problema con el factor VII. Debido a que el cuerpo produce menos factor VII del que debiera o debido a que el factor VII no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor VII es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia de factor VII es muy poco común. La deficiencia de factor VII puede heredarse junto con otras deficiencias de factor (véase la sección sobre “Deficiencia combinada de factores de la coagulación dependientes de la vitamina K”). También puede presentarse en una etapa posterior de la vida, como resultado de enfermedad hepática, deficiencia de vitamina K o consumo de ciertos medicamentos como la warfarina, un fármaco anticoagulante.
Los síntomas de la deficiencia de factor VII varían dependiendo de cada persona. Como regla general, entre menor sea la concentración de factor VII en la sangre de una persona, mayor será la frecuencia y/o gravedad de los síntomas. Las personas con concentraciones muy bajas de factor VII pueden presentar síntomas muy graves.
Las personas con deficiencia de factor VII podrían presentar hemorragias tales como las siguientes:
La deficiencia de factor VII se diagnostica mediante diversas pruebas que debería realizar un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación.
Hay varios tratamientos disponibles para la deficiencia de factor VII.
Los periodos menstruales abundantes en mujeres con deficiencia de factor VII pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia de factor X es un trastorno de la coagulación hereditario causado por un problema con el factor X. Debido a que el cuerpo produce menos factor X del que debiera o debido a que el factor X no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor X es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia de factor X es uno de los trastornos de la coagulación menos comunes.
La deficiencia de factor X también puede heredarse junto con otras deficiencias de factor (véase la sección sobre “Deficiencia combinada de factores de la coagulación dependientes de la vitamina K”).
Por lo general, entre menor sea la concentración de factor X en la sangre de una persona, mayor será la frecuencia y/o gravedad de los síntomas. Las personas con una deficiencia importante de factor X pueden presentar episodios hemorrágicos graves.
Las personas con deficiencia de factor X podrían presentar hemorragias tales como las siguientes:
La deficiencia del factor X se diagnostica mediante varias pruebas sanguíneas que debe realizar un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación.
Hay tres tratamientos disponibles para la deficiencia de factor X.
Los periodos menstruales abundantes en mujeres con deficiencia de factor X pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia combinada de factores de la coagulación dependientes de la vitamina K (VKCFD, por su sigla en inglés) es un trastorno de la coagulación hereditario muy poco común causado por un problema con los factores de coagulación II, VII, IX y X. Para que la reacción en cadena de la coagulación pueda continuar, estos cuatro factores necesitan activarse en una reacción química en la que participa la vitamina K. Cuando esta reacción no ocurre como debiera, la cascada de la coagulación se interrumpe y el coágulo sanguíneo no se forma.
La VKCFD es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. Esta deficiencia es muy poco común.
La VKCFD también puede presentarse en una etapa posterior de la vida, como resultado de trastornos gastrointestinales, enfermedad hepática, deficiencia de vitamina K o consumo de ciertos medicamentos como la warfarina, un fármaco anticoagulante. La deficiencia adquirida es más común que la deficiencia heredada. Algunos bebés recién nacidos presentan una deficiencia transitoria de vitamina K, la cual puede corregirse administrando suplementos después del nacimiento.
Los síntomas de la VKCFD varían considerablemente de una persona a otra, pero por lo general son leves. Los primeros síntomas podrían presentarse al nacimiento o en una etapa posterior de la vida. Los síntomas al nacer deben diferenciarse de la deficiencia adquirida. Las personas con deficiencias importantes pueden presentar episodios hemorrágicos graves, pero los síntomas más graves generalmente son poco comunes y solo se presentan en personas con concentraciones de factor muy bajas.
Las personas con VKCFD podrían presentar hemorragias tales como las siguientes:
La VKCFD se diagnostica mediante varias pruebas sanguíneas que debe realizar un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación. Debe tenerse cuidado, particularmente en el caso de recién nacidos, de excluir la probabilidad de una deficiencia de vitamina K adquirida o de la exposición a ciertos medicamentos.
Hay tres tratamientos disponibles para la VKCFD.
La deficiencia de factor XI es un trastorno de la coagulación hereditario causado por un problema con el factor XI. Debido a que el cuerpo produce menos factor XI del que debiera o debido a que el factor XI no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor XI también se conoce como hemofilia C. Difiere de las hemofilias A y B por el hecho de que no hay hemorragias en articulaciones y músculos. La deficiencia de factor XI es la más frecuente de las deficiencias poco comunes de factores de la coagulación y el segundo trastorno de la coagulación que más comúnmente afecta a las mujeres (después de la enfermedad de Von Willebrand).
El patrón de herencia de la deficiencia de factor XI es autosómico recesivo; sin embargo, algunas personas han heredado la deficiencia de factor XI cuando solo uno de los padres es portador del gene. Este trastorno es más común entre los judíos Askenazi (judíos de ascendencia proveniente de Europa del Este).
La mayoría de las personas con deficiencia de factor XI presentará pocos o nulos síntomas. La relación entre la cantidad de factor XI en la sangre de una persona y la gravedad de sus síntomas no es clara; personas con solo una deficiencia leve de factor XI pueden presentar episodios hemorrágicos graves. Los síntomas de la deficiencia de factor XI varían considerablemente, incluso entre miembros de una misma familia, lo cual puede dificultar su diagnóstico.
Las personas con deficiencia de factor XI podrían presentar hemorragias tales como las siguientes:
La deficiencia de factor XI se diagnostica mediante varias pruebas sanguíneas que debe realizar un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación.
Hay varios tratamientos disponibles para ayudar a controlar hemorragias en personas con deficiencia de factor XI.
Los periodos menstruales abundantes en mujeres con deficiencia de factor XI pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
La deficiencia de factor XII es un trastorno de la coagulación hereditario causado por un problema con el factor XII. Debido a que el cuerpo produce menos factor XII del que debiera o debido a que el factor XII no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor XII es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia de factor XII es muy poco común.
La mayoría de las personas con deficiencia de factor XII no presenta muchos síntomas, incluso después de cirugías mayores. Sin embargo, algunas personas presentan mala cicatrización de las heridas.
La deficiencia de factor XII a menudo se diagnostica accidentalmente durante una prueba de coagulación sanguínea de rutina realizada antes de una cirugía o debido al historial familiar. El diagnóstico se realiza mediante las pruebas de tiempo de protrombina (TP) y de tiempo de tromboplastina parcial activada (TTPa), seguidas de un ensayo de factor XII para confirmar el diagnóstico.
Las personas con deficiencia de factor XII por lo general no necesitan tratamiento dado que los síntomas habitualmente son leves o nulos. No obstante, sigue siendo importante notificar a los proveedores de atención médica o dental antes de cualquier procedimiento.
La deficiencia de factor XIII es un trastorno de la coagulación hereditario causado por un problema con el factor XIII. Debido a que el cuerpo produce menos factor XIII del que debiera o debido a que el factor XIII no funciona adecuadamente, la reacción de coagulación se interrumpe prematuramente y el coágulo sanguíneo no se forma.
La deficiencia de factor XIII es un trastorno autosómico recesivo, lo cual quiere decir que ambos padres deben ser portadores del gene defectuoso a fin de transmitirlo a sus hijos. También implica que el trastorno afecta tanto a hombres como a mujeres. La deficiencia de factor XIII es muy poco común.
La mayoría de las personas con deficiencia de factor XIII presenta síntomas desde el nacimiento, a menudo con hemorragias del muñón del cordón umbilical. Los síntomas tienden a presentarse a lo largo de toda la vida. Generalmente, entre menor sea la concentración de factor XIII en la sangre de una persona, mayor será la frecuencia y/o gravedad de los síntomas.
Las personas con deficiencia de factor XIII podrían presentar hemorragias tales como las siguientes:
La deficiencia de factor XIII es difícil de diagnosticar. Las pruebas de coagulación sanguíneas habituales no detectan la deficiencia, y muchos laboratorios no tienen a su disposición pruebas más especializadas que midan la concentración de factor XIII en una muestra de sangre o qué tan bien funciona el factor. Las pruebas debería realizarlas un especialista en un centro de tratamiento de hemofilia/trastornos de la coagulación. La elevada tasa de hemorragias al nacer generalmente resulta en un diagnóstico temprano.
Hay varios tratamientos disponibles para ayudar a controlar hemorragias en personas con deficiencia de factor XIII.
Los periodos menstruales abundantes en mujeres con deficiencia de factor XIII pueden controlarse con fármacos antifibrinolíticos o con anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] en caso de mujeres que no desean tener hijos.
Para personas con deficiencia de factor X, deficiencia de factor XIII, y afibrinogenemia, o para quienes tienen un historial de hemorragias graves y frecuentes, se recomienda el tratamiento profiláctico a largo plazo. La profilaxis es la administración (intravenosa, subcutánea o de otro tipo) periódica de un agente hemostático con el propósito de prevenir hemorragias (particularmente hemorragias que ponen en peligro la vida o hemorragias articulares recurrentes).
Aunque la tecnología se encuentra disponible, hasta el momento de esta publicación (2023) no hay ensayos en curso o terapias génicas aprobadas para las deficiencias poco comunes de factores de la coagulación.
Como ocurre con todos los medicamentos, estos tratamientos podrían tener efectos secundarios. Las personas con deficiencias poco comunes de factores de la coagulación deberían hablar con sus médicos sobre los posibles efectos secundarios del tratamiento.
Los fármacos antifibrinolíticos, ácido tranexámico y ácido aminocaproico, se utilizan para mantener un coágulo en su lugar en algunas partes del cuerpo, tales como boca, vejiga y útero. También son muy útiles en diversas situaciones; por ejemplo, durante trabajos dentales, pero no son eficaces en casos de hemorragia interna grave o cirugía. Los fármacos antifibrinolíticos son particularmente útiles para pacientes con deficiencia de factor XI. También se utilizan para ayudar a controlar el flujo menstrual excesivo. Los fármacos antifibrinolíticos pueden administrarse por vía oral o mediante inyección. Estos fármacos deberían evitarse en caso de hemorragia de las vías urinarias.
Cuando se encuentran disponibles, los concentrados de factor constituyen el tratamiento ideal y más seguro para los trastornos poco comunes. Desafortunadamente, solo hay concentrados individuales de los factores I, VII, X, XI y XIII. Los concentrados de factor para los trastornos de la coagulación poco comunes generalmente se fabrican a partir de plasma humano y reciben tratamiento a fin de eliminar virus como el VIH y los virus de la hepatitis B y C. También están disponibles el factor VIII recombinante y el factor VIIa recombinante. Estos últimos se fabrican en el laboratorio y no a partir de plasma humano, de modo que no conllevan riesgo de transmisión de enfermedades infecciosas. Los concentrados de factor se administran por vía intravenosa.
Este concentrado se fabrica a partir de plasma humano y contiene una mezcla de factores de coagulación, entre ellos los factores II, VII, IX y X (no obstante, algunos productos no contienen estos cuatro factores). El CCP es adecuado para el tratamiento de deficiencias individuales de factor II y de factor X, así como el de la deficiencia combinada de factores de la coagulación dependientes de la vitamina K (VKCFD). El concentrado es sometido a tratamiento para eliminar virus como el VIH y los de la hepatitis B y C. Hay informes de que algunos CCP podrían provocar peligrosos coágulos sanguíneos (trombosis). El CCP se administra por vía intravenosa.
El plasma es la fracción de la sangre que contiene todos los factores de la coagulación, así como otras proteínas sanguíneas. El PFC se utiliza para el tratamiento de trastornos de la coagulación poco comunes cuando los concentrados para el tratamiento del factor específico faltante no se encuentran disponibles. El PFC es el tratamiento habitual para la deficiencia de factor V. Sin embargo, generalmente no es sometido a procesos de inactivación viral, de modo que el riesgo de transmisión de enfermedades infecciosas es mayor. El PFC sometido a inactivación viral está disponible en algunos países y es preferible. La sobrecarga circulatoria constituye un problema potencial de este tratamiento: debido a que en el PFC la concentración de cada factor de coagulación es baja, debe administrarse un volumen considerable durante varias horas a fin de elevar adecuadamente la concentración del factor faltante. Esta gran cantidad de PFC puede sobrecargar al sistema circulatorio y forzar al corazón. El tratamiento con PFC puede presentar otras complicaciones, particularmente reacciones alérgicas o problemas pulmonares (lesiones pulmonares provocadas por transfusiones sanguíneas). El riesgo de estas lesiones puede reducirse evitando la transfusión de PFC recolectado de mujeres que tuvieron embarazos previos. Estos problemas son mucho menos comunes si se utilizan lotes de PFC sometidos a inactivación viral o PFC con patógenos reducidos. El PFC se administra por vía intravenosa.
Fabricado a partir de plasma humano, el crioprecipitado contiene factor VIII, fibrinógeno (factor I) y algunas otras proteínas importantes para la coagulación de la sangre. El crioprecipitado sin patógenos reducidos no es sometido a inactivación viral y solo debería utilizarse cuando el concentrado de factor no se encuentre disponible. Los centros de transfusión sanguínea pueden preparar crioprecipitado con patógenos reducidos o sometido a inactivación viral, los cuales deberían usarse cuando no haya CFC disponibles. En comparación con el PFC, el crioprecipitado contiene mayores concentraciones de algunos de los factores de la coagulación (pero no de todos), de modo que el volumen requerido es menor. Solo es adecuado para unas cuantas deficiencias. El crioprecipitado se administra por vía intravenosa.
La desmopresina (DDAVP) es una hormona sintética que eleva las concentraciones de factor VIII en pacientes con deficiencia combinada de factor V y factor VIII. Dado que es sintética, no hay riesgo de transmisión de enfermedades infecciosas. La desmopresina no afecta las concentraciones de ninguno de los otros factores de coagulación. Puede administrarse por vía intranasal, intravenosa o subcutánea.
La goma o cola de fibrina puede usarse para el tratamiento de lesiones externas y durante procedimientos odontológicos tales como extracciones dentales. No se utiliza en caso de hemorragias mayores o cirugías. Se aplica directamente al sitio de la hemorragia.
Las plaquetas son pequeñas células sanguíneas que participan en la formación de coágulos y en la reparación de vasos sanguíneos lesionados. Algunos concentrados de factor, entre ellos el factor V, se almacenan en pequeños sacos al interior de las plaquetas. Las transfusiones de plaquetas algunas veces se utilizan para el tratamiento de la deficiencia de factor V.
El tratamiento con vitamina K (ya sean tabletas o inyecciones) puede ayudar a controlar síntomas de la deficiencia combinada de los factores de la coagulación dependientes de la vitamina K (VKCFD). Sin embargo, no todas las personas responden a este tipo de tratamiento. Las personas que no responden a la vitamina K y presentan una hemorragia o requieren cirugía necesitarán terapia con factor de reemplazo.
Los anticonceptivos hormonales [tales como pastillas anticonceptivas o dispositivos/sistemas intrauterinos (DIU/SIU) liberadores de levonorgestrel] pueden ayudar a controlar periodos menstruales abundantes en mujeres que no desean tener hijos.
Saber que usted o un miembro de su familia padece un trastorno de la coagulación podría resultar muy perturbador y podría experimentar una gama de emociones diferentes. A algunas personas, el diagnóstico podría causarles miedo y ansiedad, mientras que, para otras, poder darle un nombre a los síntomas con los que han vivido durante años podría ser un gran alivio. Los padres podrían sentirse culpables de saber que su hijo(a) ha heredado un trastorno genético. Todos estos sentimientos son normales y es probable que cambien con el transcurso del tiempo, conforme usted aprenda más sobre el trastorno y el impacto que tendrá en su vida y la de su(s) familiar(es).
Hablar con otras personas –amigos, padres, profesionales de la salud y otros pacientes con trastornos de la coagulación– puede resultar muy consolador. Aprender tanto como pueda acerca del trastorno también le ayudará a sentir más confianza y a calmar sus miedos. Comuníquese con la organización de pacientes o centro de tratamiento de hemofilia/trastornos de la coagulación de su comunidad para hacer preguntas y hablar sobre sus opciones. Puede ubicar grupos de pacientes y centros de tratamiento usando la página internet de la FMH, en wfh.org/es/encuentre-apoyo-local/.
Las personas con trastornos de la coagulación deberían recibir seguimiento en un centro de tratamiento especializado en el diagnóstico y tratamiento de estos trastornos, ya que es probable que dichos centros sean los que ofrezcan las mejores normas de atención e información.
Una dieta sana y el ejercicio cotidiano contribuyen a mantener al cuerpo saludable y fuerte. El ejercicio también puede ayudar a disminuir el estrés, la ansiedad y la depresión, así como la frecuencia y gravedad de hemorragias articulares.
Las personas con trastornos de la coagulación graves que corren el riesgo de padecer hemorragias articulares deberían evitar actividades y deportes de alto impacto, tales como fútbol, luchas y patineta. Idealmente, los ejercicios para las personas con trastornos de la coagulación debería recomendarlos un médico o fisioterapeuta calificado y con experiencia.
Una buena higiene oral es indispensable a fin de evitar caries dentales y enfermedad de las encías. Para las personas con trastornos de la coagulación es muy importante mantener una buena salud bucal con el objeto de disminuir la necesidad de cirugías dentales que podrían complicarse por hemorragias excesivas o prolongadas. Las personas con trastornos de la coagulación deberían hacer lo siguiente:
Procedimientos invasores, tales como raspado de raíces, extracciones dentales o endodoncias, podrían provocar hemorragias en personas con trastornos de la coagulación. Incluso en caso de procedimientos menores, el dentista debería consultar al centro de tratamiento de trastornos de la coagulación a fin de determinar los posibles riesgos que corre el paciente y de elaborar un plan adecuado para prevenir o controlar hemorragias en cualquier tipo de procedimiento. Podría ser necesario administrar medicamentos antes de la intervención a fin de prevenir hemorragias y garantizar un procedimiento y una recuperación sin complicaciones.
Las personas con trastornos de la coagulación deberían vacunarse. Las vacunas deben administrarse por vía subcutánea (bajo la piel) y no directamente al músculo, a fin de evitar el riesgo de hemorragia correspondiente, y esto debería conversarse con el médico que atiende el trastorno de la coagulación.
Las vacunas contra las hepatitis A y B son particularmente importantes para personas que reciben tratamiento con plasma fresco congelado y con cualquier otro producto que no haya sido sometido a inactivación viral. Los familiares que ayudan a administrar tratamiento también deberían vacunarse, aunque esto es menos importante en el caso de personas que utilizan productos sometidos a inactivación viral.
Verifique todos los medicamentos con su médico. Algunas medicinas vendidas sin receta deben evitarse porque interfieren con la coagulación. Las personas con trastornos de la coagulación no deberían tomar aspirina (ácido acetilsalicílico) o fármacos antiinflamatorios no esteroides (como ibuprofeno y naproxeno) sin asesoría médica.
Las personas con trastornos de la coagulación siempre deberían llevar consigo información sobre su trastorno, el tratamiento necesario, y el nombre y número telefónico de su médico o centro de tratamiento. En casos de emergencia, un brazalete de alerta médica u otra identificación, como la tarjeta médica internacional de la FMH, notifica al personal de salud sobre su trastorno de la coagulación. Antes de viajar busque las direcciones y números telefónicos de los centros de tratamiento de trastornos de la coagulación de su(s) lugar(es) de destino y lleve la información consigo, en caso de que necesitara atención médica. La información de los centros de tratamiento puede encontrarse en la página internet de la FMH, en wfh.org/es/encuentre-apoyo-local/.
Las mujeres con deficiencias de factores de la coagulación podrían presentar más síntomas que los hombres debido al riesgo de hemorragias relacionadas con la menstruación y el parto. Las niñas podrían presentar flujos abundantes al inicio de su menstruación.
Las mujeres con deficiencias de factores de la coagulación podrían tener flujos menstruales más abundantes y/o prolongados, lo cual podría provocar una deficiencia de hierro (bajos niveles de hierro que causan debilidad y cansancio) y/o anemia (bajas concentraciones de glóbulos rojos en la sangre).
Antes de planear cualquier embarazo, las mujeres con deficiencias de factores de la coagulación deberían recibir asesoría genética sobre los riesgos de tener un hijo que herede el trastorno, y deberían consultar a un obstetra en cuanto sospechen que pudieran estar embarazadas. El obstetra debería trabajar en estrecha colaboración con el personal del centro de tratamiento de trastornos de la coagulación a fin de ofrecer la mejor atención posible durante el embarazo y el parto, así como para reducir al mínimo posibles complicaciones tanto para la madre como para el recién nacido.
Las mujeres con ciertas deficiencias de factor (como deficiencia de factor X, deficiencia de factor XIII, y afibrinogenemia) pueden correr mayores riesgos de aborto y desprendimiento de placenta (la separación prematura de la placenta del útero, lo cual interrumpe el flujo de sangre y oxígeno al feto). Debido a lo anterior, estas mujeres requerirán tratamiento durante todo el embarazo a fin de evitar tales complicaciones.
El principal riesgo relacionado con el embarazo es la hemorragia posparto. Los trastornos de la coagulación están relacionados con un mayor riesgo de hemorragia, tanto inmediatamente como varias semanas después del parto. Por ende, las mujeres con trastornos de la coagulación deberían colaborar con sus médicos (tanto con el especialista en trastornos de la coagulación como con el obstetra) a fin de establecer un plan de parto personalizado. Dicho plan debería abordar todas las etapas del alumbramiento, inclusive la expulsión de la placenta, con el propósito de reducir el riesgo y la gravedad de la hemorragia. El tratamiento será diferente para cada mujer y dependerá de su historial personal y familiar de síntomas hemorrágicos, del diagnóstico y la gravedad de la deficiencia de factor, y del tipo de parto. Debería recomendarse a las mujeres con trastornos de la coagulación que se comuniquen con su proveedor de atención médica inmediatamente si la hemorragia posparto fuera excesiva.
En algunas circunstancias, los bebés nacidos de mujeres con trastornos de la coagulación podrían también correr el riesgo de heredar el trastorno y presentar hemorragias. Deberían evitarse el trabajo de parto difícil y prolongado, y alumbramientos con instrumentos, tales como el uso de fórceps o extracción con ventosas.






