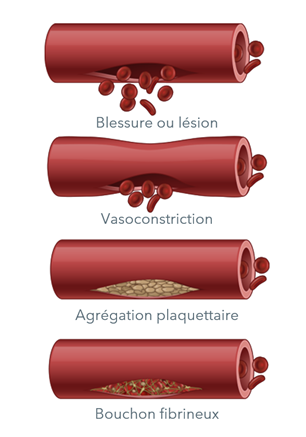
Les facteurs de coagulation sont des protéines présentes dans le sang qui contrôlent les saignements. Lorsqu’un vaisseau sanguin est endommagé, ses parois se contractent pour limiter le flux sanguin vers la zone lésée. Ensuite, de petites cellules sanguines appelées plaquettes se fixent sur le site de la blessure et se répandent à la surface du vaisseau sanguin pour arrêter le saignement. Parallèlement à cela, de petits sacs situés à l’intérieur des plaquettes libèrent des substances chimiques qui attirent d’autres plaquettes vers la zone lésée et les font s’agglutiner pour former ce que l’on appelle un clou plaquettaire.
À la surface de ces plaquettes activées, de nombreux facteurs de coagulation différents agissent en synergie dans une série de réactions chimiques complexes (connues sous le nom de cascade de la coagulation) pour former un caillot de fibrine. Le caillot agit comme un filet pour arrêter le saignement.
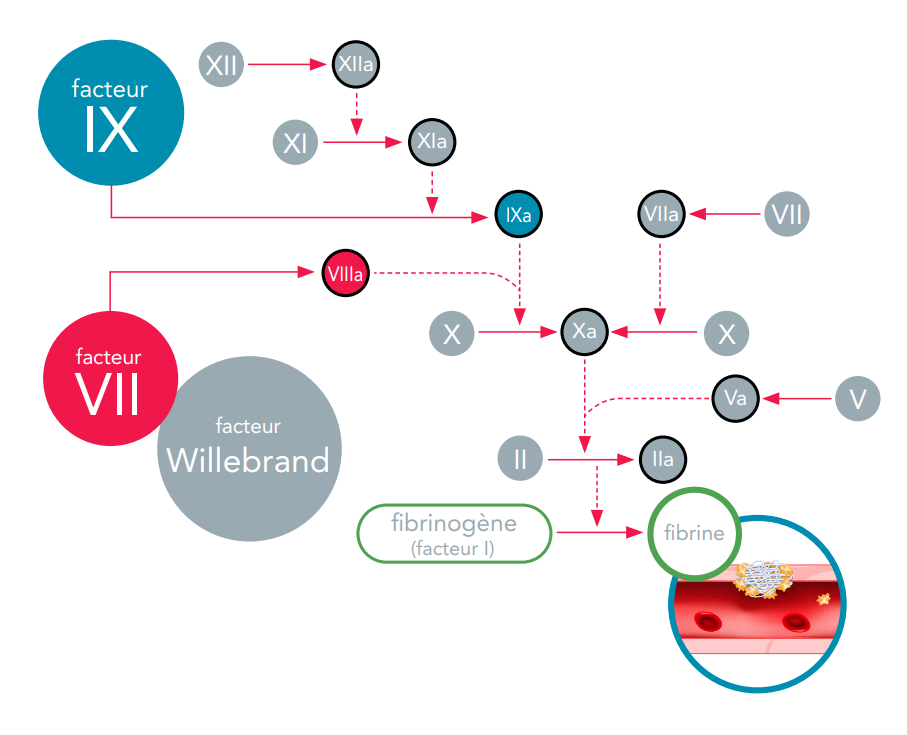
Les facteurs de coagulation circulent dans le sang sous une forme inactive. Lorsqu’un vaisseau sanguin est lésé, la cascade de la coagulation est déclenchée et chaque facteur de coagulation est activé dans un ordre précis pour conduire à la formation du caillot sanguin. Les facteurs de coagulation sont identifiés par des chiffres romains (par exemple, facteur VIII ou FVIII). L’ajout de la lettre a (par exemple, facteur VIIIa) indique la forme activée du facteur de coagulation.
La cascade de la coagulation est une série de voies qui convergent vers le facteur X, facteur de coagulation essentiel à la libération de la fibrine et à la formation du caillot.
Chez les personnes atteintes d’un déficit ou, plus rarement, de plusieurs déficits en facteur de coagulation, le facteur de coagulation concerné est absent, est présent à un faible niveau ou ne fonctionne pas correctement. Cela a une incidence sur le processus normal de coagulation du sang, rendant difficile la formation d’un caillot.
En cas d’absence ou de dysfonctionnement d’un facteur de coagulation, la cascade de la coagulation est bloquée. Il en découle que le caillot sanguin ne se forme pas, et l’hémorragie se prolonge alors plus longtemps qu’elle ne le devrait.
Les déficits en facteur VIII et en facteur IX sont connus sous le nom d’hémophilie A et B, respectivement. Le déficit ou l’anomalie du facteur Willebrand est connu sous le nom de maladie de Willebrand. Les déficits rares en facteur de coagulation sont des troubles de la coagulation pour lesquels un ou plusieurs des autres facteurs de coagulation (c’est-à-dire les facteurs I, II, V, V+VIII, VII, X, XI, XII ou XIII) sont absents ou ne fonctionnent pas correctement. Ces troubles sont peu connus, car ils sont rarement diagnostiqués. Tous les déficits en facteur de coagulation sont des maladies rares, et les déficits rares en facteur de coagulation sont considérés comme des maladies ultra-rares, car ceux-ci touchent une population encore plus restreinte.
L’être humain possède 23 paires de chromosomes : 22 paires de chromosomes autosomiques (également appelés autosomes) et 1 paire de chromosomes sexuels (X ou Y). Contrairement à l’hémophilie, qui est due à des mutations sur le chromosome X, les déficits rares en facteur de coagulation sont dus à des mutations sur les gènes autosomes.
Le déficit en facteur I (fibrinogène) est un trouble héréditaire de la coagulation causé par un problème avec le facteur I. Lorsque l’organisme ne produit pas suffisamment de fibrinogène ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas. Le déficit en facteur I est un terme générique qui englobe plusieurs troubles apparentés, connus sous le nom d’anomalies congénitales du fibrinogène.
L’afibrinogénémie est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant.
L’hypofibrinogénémie, la dysfibrinogénémie et l’hypodysfibrinogénémie peuvent être récessives (les deux parents sont porteurs du gène) ou dominantes (un seul parent est porteur du gène et le transmet).
Tous les types de déficit en facteur I affectent aussi bien les hommes que les femmes.
Les symptômes du déficit en facteur I diffèrent selon la forme de la maladie dont la personne est atteinte.
Le fibrinogène étant un acteur central de la coagulation, son absence totale entraîne de graves troubles hémostatiques, allant de la thrombose à l’hémorragie engageant le pronostic vital. Les saignements sont les symptômes les plus fréquents. En règle générale, ils sont spontanés et peuvent prendre la forme d’hémorragies graves engageant le pronostic vital.
Les personnes atteintes d’afibrinogénémie peuvent présenter les saignements suivants :
*L’incidence des événements thrombotiques est plus élevée chez les jeunes enfants.
Les symptômes ressemblent à ceux de l’afibrinogénémie. En règle générale, plus la quantité de facteur I dans le sang d’une personne est faible, plus les symptômes sont fréquents et/ou graves.
Les symptômes dépendent du fonctionnement du fibrinogène (présent en quantité normale). Alors que certaines personnes ne présentent aucun symptôme, d’autres présentent des saignements (comme ceux observés en cas d’afibrinogénémie), et d’autres encore présentent des signes de thrombose (caillots sanguins anormaux dans les vaisseaux sanguins) au lieu de saigner.
Les symptômes sont variables et dépendent de la quantité de fibrinogène produite et du fonctionnement de celui-ci.
Le déficit en facteur I est diagnostiqué grâce à différents examens sanguins, dont un test spécifique qui mesure la quantité de fibrinogène dans le sang. Cependant, un faible taux de fibrinogène ou une fonction anormale de celui-ci peut être le signe d’une autre maladie, telle qu’un trouble hépatique ou rénal, qu’il convient d’écarter avant de diagnostiquer un trouble de la coagulation. Les tests doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation.
Le déficit en facteur I se traite de trois façons différentes. Tous ces traitements sont fabriqués à partir de plasma humain.
Il est également possible d’administrer un traitement pour prévenir la formation de caillots sanguins, car cette complication peut survenir après l’administration d’un traitement de remplacement du fibrinogène.
Beaucoup de personnes atteintes d’hypofibrinogénémie ou de dysfibrinogénémie n’ont pas besoin de traitement. Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur I peuvent être contrôlés grâce à des médicaments antifibrinolytiques, ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit en facteur II (prothrombine) est un trouble héréditaire de la coagulation causé par un problème avec le facteur II. Lorsque l’organisme ne produit pas suffisamment de prothrombine ou que celle-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur II est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit en facteur II est très rare.
Le déficit en facteur II peut être associé à d’autres déficits en facteur (voir « Déficit combiné en facteurs de coagulation dépendants de la vitamine K »). Il peut également être acquis ultérieurement, à la suite d’une maladie hépatique, d’une carence en vitamine K ou de la prise de certains médicaments tels que la warfarine, un anticoagulant. Le déficit en facteur II acquis est plus fréquent que la forme héréditaire.
Les symptômes du déficit en facteur II varient d’une personne à l’autre. En règle générale, plus la quantité de facteur II dans le sang d’une personne est faible, plus les symptômes sont fréquents et/ou graves.
Les personnes atteintes d’un déficit en facteur II peuvent présenter les saignements suivants :
Le déficit en facteur II est diagnostiqué grâce à différents examens sanguins. Le médecin doit mesurer le temps de prothrombine (TP) et la thromboplastine partielle activée (TPA). Si les deux tests présentent un temps prolongé, il doit alors mesurer le taux des facteurs I, II, V, VII, IX et X dans le sang. Les tests doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation.
Le déficit en facteur II se traite de deux façons différentes. Tous ces traitements sont fabriqués à partir de plasma humain.
Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur II peuvent être contrôlés grâce à des médicaments antifibrinolytiques, ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit en facteur V est un trouble héréditaire de la coagulation causé par un problème avec le facteur V. Lorsque l’organisme ne produit pas suffisamment de facteur V ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur V est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit en facteur V est très rare.
Les symptômes du déficit en facteur V sont généralement légers et certaines personnes peuvent n’en présenter aucun. Cependant, les enfants atteints d’un déficit sévère en facteur V peuvent présenter des hémorragies à un très jeune âge. Certaines personnes atteintes d’un déficit en facteur V peuvent connaître des saignements dans le système nerveux central (le cerveau et la moelle épinière) très tôt dans leur vie.
Les personnes atteintes d’un déficit en facteur V peuvent présenter les saignements suivants :
Le déficit en facteur V est diagnostiqué grâce à différents examens sanguins qui doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation. Le médecin doit mesurer le temps de prothrombine (TP) et la thromboplastine partielle activée (TPA). Si les deux tests présentent un temps prolongé, il doit alors mesurer le taux des facteurs I, II, V, VII, IX et X dans le sang. Les personnes présentant des taux anormaux de facteur V doivent également faire vérifier leur taux de facteur VIII afin d’exclure un déficit combiné en facteur V et en facteur VIII, un trouble tout à fait distinct.
En règle générale, il n’est nécessaire de traiter un déficit en facteur V qu’en cas d’hémorragie grave ou préalablement à une intervention chirurgicale. Le plasma frais congelé (PFC) ou le PFC réduit en agents pathogènes est le traitement habituel, car il n’existe pas de concentré contenant uniquement du facteur V. Les transfusions de plaquettes, qui contiennent du facteur V, constituent parfois également une option thérapeutique.
Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur V peuvent être contrôlés grâce à des médicaments antifibrinolytiques, ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit combiné en facteur V et en facteur VIII est un trouble héréditaire de la coagulation causé par un faible taux de facteurs V et VIII. Lorsque la quantité de ces facteurs dans l’organisme est inférieure à la normale, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas. Le déficit combiné en facteur V et en facteur VIII est un trouble tout à fait distinct du déficit en facteur V ou du déficit en facteur VIII (hémophilie A).
Le déficit combiné en facteur V et en facteur VIII est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit est très rare et il est également très rare que le déficit en facteur VIII soit hérité séparément d’un seul parent.
Dans la plupart des cas, la maladie est causée par une anomalie génétique unique qui affecte la capacité de l’organisme à transporter le facteur V et le facteur VIII en dehors de la cellule et dans la circulation sanguine, et non par un problème avec le gène de l’un ou l’autre facteur.
Le déficit combiné en facteur V et en facteur VIII ne semble pas provoquer plus d’hémorragies que si l’un ou l’autre des facteurs était atteint. Les symptômes du déficit combiné en facteur V et en facteur VIII sont généralement légers.
Les personnes atteintes d’un déficit combiné en facteur V et en facteur VIII peuvent présenter les saignements suivants :
Le déficit combiné en facteur V et en facteur VIII est diagnostiqué grâce à différents examens sanguins visant à déterminer si le taux des deux facteurs est inférieur à la normale. Les tests doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation.
Le déficit combiné en facteur V et en facteur VIII se traite de trois façons différentes.
Les saignements menstruels excessifs chez les femmes ayant un déficit combiné en facteur V et en facteur VIII peuvent être contrôlés grâce à des médicaments antifibrinolytiques ou à des contraceptifs hormonaux (tels que la pilule contraceptive, ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit en facteur VII est un trouble héréditaire de la coagulation causé par un problème avec le facteur VII. Lorsque l’organisme ne produit pas suffisamment de facteur VII ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur VII est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit en facteur VII est très rare. Il peut être associé à d’autres déficits en facteur (voir « Déficit combiné en facteurs de coagulation dépendants de la vitamine K »). Il peut également être acquis ultérieurement, à la suite d’une maladie hépatique, d’une carence en vitamine K ou de la prise de certains médicaments tels que la warfarine, un anticoagulant.
Les symptômes du déficit en facteur VII varient d’une personne à l’autre. En règle générale, plus la quantité de facteur VII dans le sang d’une personne est faible, plus les symptômes sont fréquents et/ou graves. Les personnes dont le taux de facteur VII est très bas peuvent présenter des symptômes très graves.
Les personnes atteintes d’un déficit en facteur VII peuvent présenter les saignements suivants :
Le déficit en facteur VII est diagnostiqué grâce à différents examens sanguins qui doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation.
Le déficit en facteur VII se traite de plusieurs façons.
Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur VII peuvent être contrôlés grâce à des médicaments antifibrinolytiques ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit en facteur X est un trouble héréditaire de la coagulation causé par un problème avec le facteur X. Lorsque l’organisme ne produit pas suffisamment de facteur X ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur X est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit en facteur X est un des plus rare troubles de la coagulation.
Le déficit en facteur X peut être associé à d’autres déficits en facteur (voir « Déficit combiné en facteurs de coagulation dépendants de la vitamine K »).
En règle générale, plus la quantité de facteur X dans le sang d’une personne est faible, plus les symptômes sont fréquents et/ou graves. Les personnes atteintes d’un déficit en facteur X sévère peuvent présenter des épisodes hémorragiques graves.
Les personnes atteintes d’un déficit en facteur X peuvent présenter les saignements suivants :
Le déficit en facteur X est diagnostiqué grâce à différents examens sanguins qui doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation.
Le déficit en facteur X se traite de trois façons différentes.
Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur X peuvent être contrôlés grâce à des médicaments antifibrinolytiques ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K est un trouble hémorragique très rare causé par un problème avec les facteurs de coagulation II, VII, IX et X. Pour permettre la réaction en chaîne de la cascade de la coagulation, ces quatre facteurs doivent être activés dans une réaction chimique qui implique la vitamine K. Lorsque cette réaction ne se produit pas comme elle le devrait, la cascade de la coagulation est bloquée, et le caillot sanguin ne se forme pas.
Le déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K est très rare.
Le déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K peut également être acquis ultérieurement, à la suite de troubles intestinaux, d’une maladie hépatique, d’une carence alimentaire en vitamine K ou de la prise de certains médicaments tels que la warfarine, un anticoagulant. La carence en vitamine K acquise est plus fréquente que la forme héréditaire. Certains nouveau-nés présentent une carence temporaire en vitamine K, laquelle peut être traitée par des suppléments à la naissance.
Les symptômes du déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K varient considérablement d’une personne à l’autre, mais sont généralement légers. Les premiers symptômes peuvent apparaître dès la naissance ou bien plus tard. Les symptômes à la naissance doivent être différenciés des symptômes du déficit acquis. Les personnes atteintes de déficits sévères peuvent présenter des épisodes hémorragiques graves, mais les symptômes les plus graves sont généralement rares et ne surviennent que chez les personnes dont le taux de facteur est très bas.
Les personnes atteintes d’un déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K peuvent présenter les saignements suivants :
Le déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K est diagnostiqué grâce à différents examens sanguins qui doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation. Il convient, notamment chez les nouveau‑nés, d’exclure les causes d’une carence en vitamine K acquise ou d’éviter de les exposer à certains médicaments.
Le déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K se traite de trois façons différentes.
Le déficit en facteur XI est un trouble héréditaire de la coagulation causé par un problème avec le facteur XI. Lorsque l’organisme ne produit pas suffisamment de facteur XI ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur XI est également appelé hémophilie C. Elle diffère des hémophilies A et B par l’absence de saignement dans les articulations et les muscles. Le déficit en facteur XI est le plus fréquent des déficits rares en facteur de coagulation et le deuxième trouble de la coagulation le plus fréquent chez les femmes (après la maladie de Willebrand).
Le déficit en facteur XI est une maladie autosomique récessive ; toutefois, il peut arriver que certaines personnes en héritent lorsqu’un seul de leurs parents est porteur du gène. La maladie est plus fréquente chez les juifs ashkénazes (juifs d’origine de l’Europe de l’est).
La plupart des personnes atteintes d’un déficit en facteur XI ne présentent que peu ou pas de symptômes. La relation entre le taux de facteur XI dans le sang d’une personne et la gravité de ses symptômes n’est pas claire ; les personnes qui ne présentent qu’un léger déficit en facteur XI peuvent avoir de graves épisodes hémorragiques. Les symptômes du déficit en facteur XI varient considérablement, même entre les membres d’une même famille, ce qui peut rendre le diagnostic difficile.
Les personnes atteintes d’un déficit en facteur XI peuvent présenter les saignements suivants :
Le déficit en facteur XI est diagnostiqué grâce à différents examens sanguins qui doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation.
Le déficit en facteur XI se traite de plusieurs façons.
Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur XI peuvent être contrôlés grâce à des médicaments antifibrinolytiques ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Le déficit en facteur XII est un trouble héréditaire de la coagulation causé par un problème avec le facteur XII. Lorsque l’organisme ne produit pas suffisamment de facteur XII ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur XII est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit en facteur XII est très rare.
La plupart des personnes atteintes d’un déficit en facteur XII ne présentent pas beaucoup de symptômes, même après une intervention chirurgicale majeure. Cependant, certaines ont des problèmes de cicatrisation.
Le déficit en facteur XII est souvent diagnostiqué accidentellement, lors d’une analyse sanguine de routine pour vérifier la coagulation avant une intervention chirurgicale ou en raison d’antécédents familiaux. Le diagnostic est établi en déterminant le temps de prothrombine (TP) et la thromboplastine partielle activée (TPA), qui seront suivis d’un dosage du facteur XII pour confirmer le diagnostic.
Les personnes atteintes d’un déficit en facteur XII n’ont généralement pas besoin de traitement, car les symptômes sont habituellement légers ou inexistants. Toutefois, il est important d’en informer les professionnels de santé ou les dentistes avant toute intervention.
Le déficit en facteur XIII est un trouble héréditaire de la coagulation causé par un problème avec le facteur XIII. Lorsque l’organisme ne produit pas suffisamment de facteur XIII ou que celui-ci ne fonctionne pas correctement, la cascade de la coagulation est bloquée prématurément, et le caillot sanguin ne se forme pas.
Le déficit en facteur XIII est une maladie autosomique récessive, ce qui signifie que les deux parents doivent être porteurs du gène défectueux pour le transmettre à leur enfant. Cela signifie également que la maladie touche aussi bien les hommes que les femmes. Le déficit en facteur XIII est très rare.
La plupart des personnes atteintes d’un déficit en facteur XIII présentent des symptômes dès la naissance, souvent un saignement du cordon ombilical. Les symptômes ont tendance à persister tout au long de la vie. En règle générale, plus la quantité de facteur XIII dans le sang d’une personne est faible, plus les symptômes sont fréquents et/ou graves.
Les personnes atteintes d’un déficit en facteur XIII peuvent présenter les saignements suivants :
Le déficit en facteur XIII est difficile à diagnostiquer. Les tests de coagulation standard ne permettent pas de détecter ce déficit, et de nombreux laboratoires ne possèdent pas les équipements nécessaires pour effectuer des tests plus spécialisés destinés à mesurer le taux de facteur XIII dans un échantillon de sang ou l’efficacité du facteur XIII. Les tests doivent être effectués par un spécialiste dans un centre de traitement de l’hémophilie et des troubles de la coagulation. Le taux élevé de saignements à la naissance permet généralement un diagnostic précoce.
Le déficit en facteur XIII se traite de plusieurs façons.
Les saignements menstruels excessifs chez les femmes ayant un déficit en facteur XIII peuvent être contrôlés grâce à des médicaments antifibrinolytiques ou à des contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) chez les femmes ne souhaitant pas avoir d’enfant.
Chez les personnes atteintes d’un déficit en facteur X, d’un déficit en facteur XIII ou d’une afibrinogénémie, ou chez celles qui ont des antécédents de saignements graves et fréquents, il est recommandé de mettre en œuvre une prophylaxie à long terme. La prophylaxie est l’administration régulière (par voie intraveineuse ou sous‑cutanée, par exemple) d’un agent hémostatique dans le but de prévenir les saignements (notamment les saignements engageant le pronostic vital ou les saignements articulaires récurrents).
Au moment de la publication (2023) de la présente brochure, aucun essai clinique n’est en cours ni aucune thérapie génique n’est approuvée pour les déficits rares en facteur de coagulation, et ce, malgré l’existence d’une telle technologie dans le domaine de l’hémophilie.
Comme tous les médicaments, ces traitements peuvent entraîner des effets secondaires. Les personnes atteintes d’un déficit rare en facteur de coagulation doivent parler à leur médecin des effets secondaires possibles du traitement.
Les médicaments antifibrinolytiques, comme l’acide tranexamique ou l’acide aminocaproïque, visent à maintenir un caillot en place dans certaines parties du corps, telles que la bouche, la vessie et l’utérus. Ils sont également très utiles dans de nombreuses situations, par exemple dans le cadre de soins dentaires, mais ne sont pas efficaces en cas d’hémorragie interne importante ou d’intervention chirurgicale. Les médicaments antifibrinolytiques sont particulièrement utiles pour les personnes atteintes d’un déficit en facteur XI. Ils sont également utilisés pour aider à contrôler les saignements menstruels excessifs. Les médicaments antifibrinolytiques peuvent être administrés par voie orale ou par injection. Il est recommandé d’éviter tout médicament antifibrinolytique en cas d’hémorragie urinaire.
Lorsqu’ils sont disponibles, les concentrés de facteur constituent le traitement idéal et le plus sûr pour les troubles rares de la coagulation. Malheureusement, les concentrés individuels ne sont disponibles que pour les facteurs I, VII, X, XI et XIII. Les concentrés de facteur pour les troubles rares de la coagulation sont généralement fabriqués à partir de plasma humain et sont traités pour éliminer les virus tels que le VIH et les hépatites B et C. Le facteur VIII recombinant et le facteur VIIa recombinant sont également disponibles. Ils sont fabriqués en laboratoire et non à partir de plasma humain, et ne présentent donc aucun risque de maladie infectieuse. Les concentrés de facteur sont administrés par voie intraveineuse.
Ce concentré est fabriqué à partir de plasma humain et contient un mélange de facteurs de coagulation, notamment les facteurs II, VII, IX et X (certains produits ne contiennent toutefois pas les quatre facteurs). Le concentré de complexe prothrombique convient aux déficits individuels en facteurs II et X ainsi qu’au déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K. Il est traité pour éliminer les virus tels que le VIH et les hépatites B et C. Il a été rapporté que certains concentrés de complexe prothrombique provoqueraient des caillots sanguins potentiellement dangereux (thrombose). Le concentré de complexe prothrombique est administré par voie intraveineuse.
Le plasma est la partie du sang qui contient tous les facteurs de coagulation, ainsi que d’autres protéines sanguines. Le plasma frais congelé est utilisé pour traiter des troubles rares de la coagulation lorsque les concentrés du facteur manquant ou déficient ne sont pas disponibles. Le plasma frais congelé est le traitement habituel en cas de déficit en facteur V. Cependant, il ne subit généralement pas d’inactivation virale, ce qui augmente le risque de transmission de maladies infectieuses. Le plasma frais congelé viro-inactivé est disponible dans certains pays et doit être privilégié. La surcharge circulatoire est un problème potentiel de ce traitement : comme la concentration de chaque facteur de coagulation dans le plasma frais congelé est faible, il faut en administrer un grand volume pendant plusieurs heures pour obtenir une augmentation adéquate du taux de facteur. Cette grande quantité de plasma frais congelé peut surcharger le système circulatoire et stresser le cœur. D’autres complications dans le cadre du traitement par plasma frais congelé peuvent survenir, en particulier des réactions allergiques ou des problèmes pulmonaires (lésions pulmonaires liées à la transfusion). Le risque de lésions pulmonaires liées à la transfusion peut être réduit en évitant de transfuser du plasma frais congelé prélevé chez une femme ayant déjà été enceinte. Ces problèmes sont beaucoup moins fréquents si l’on utilise du pool de plasma frais congelé viro-inactivé ou du plasma frais congelé réduit en agents pathogènes. Le plasma frais congelé est administré par voie intraveineuse.
Fabriqué à partir de plasma humain, le cryoprécipité contient du facteur VIII, du fibrinogène (facteur I) et quelques autres protéines importantes pour la coagulation sanguine. Le cryoprécipité sans réduction des agents pathogènes ne subit pas d’inactivation virale et ne doit être utilisé que lorsque le concentré de facteur concerné n’est pas disponible. Le cryoprécipité réduit en agents pathogènes ou ayant fait l’objet d’une inactivation virale peut être préparé par les centres de transfusion sanguine et doit être utilisé en l’absence de concentrés de facteur de coagulation. Il contient des concentrations plus élevées de certains facteurs de coagulation (mais pas tous) que le plasma frais congelé, de sorte qu’il est possible d’en administrer un volume moindre. Il ne convient que pour quelques déficits. Le cryoprécipité est administré par voie intraveineuse.
La desmopressine (DDAVP) est une hormone synthétique qui augmente le taux de facteur VIII chez les patients présentant un déficit combiné en facteur V et en facteur VIII. Comme elle est synthétique, il n’y a aucun risque de transmission de maladies infectieuses. La desmopressine n’a aucun effet sur le taux des autres facteurs de coagulation. Elle peut être administrée par voie intranasale, intraveineuse ou sous-cutanée.
La colle de fibrine peut être utilisée pour traiter les plaies externes et lors de soins dentaires, comme l’extraction d’une dent. Elle n’est pas utilisée en cas d’hémorragie importante ou d’intervention chirurgicale. Elle est appliquée directement sur le site de l’hémorragie.
Les plaquettes sont de petites cellules sanguines qui participent à la formation de caillots sanguins et à la réparation des vaisseaux sanguins endommagés. Certains facteurs de coagulation, dont le facteur V, sont stockés dans de petits sacs à l’intérieur des plaquettes. Les transfusions de plaquettes sont parfois utilisées pour traiter le déficit en facteur V.
Un traitement à la vitamine K (sous forme de comprimés ou par injection) peut aider à contrôler les symptômes d’un déficit combiné héréditaire en facteurs de coagulation dépendants de la vitamine K. Cependant, tout le monde ne répond pas à ce type de traitement. Les personnes qui ne répondent pas à la vitamine K et qui ont un saignement ou doivent subir une intervention chirurgicale auront besoin d’un facteur de remplacement.
Les contraceptifs hormonaux (tels que la pilule contraceptive ou le dispositif intra-utérin à libération de lévonorgestrel) peuvent aider à contrôler les saignements menstruels chez les femmes ne souhaitant pas avoir d’enfant.
Apprendre que vous ou votre enfant êtes atteints d’un trouble de la coagulation peut être bouleversant et cette annonce peut vous faire vivre toute une gamme d’émotions. Certaines personnes peuvent ressentir de la peur et de l’anxiété, alors que pour d’autres, le fait de pouvoir mettre un nom sur leurs symptômes s’avère un énorme soulagement. Les parents peuvent se sentir coupables en apprenant que leur enfant a hérité d’une maladie génétique. Tous ces sentiments sont normaux et sont susceptibles d’évoluer au fil du temps, à mesure que vous en apprendrez davantage sur la maladie et sur l’incidence qu’elle aura sur votre vie ou celle d’un membre de votre famille.
Il peut être réconfortant d’échanger avec d’autres personnes (amis, parents, professionnels de santé et autres personnes atteintes de troubles de la coagulation). Le fait d’en apprendre le plus possible sur la maladie vous aidera également à vous sentir plus confiant et à dissiper vos craintes. Entrez en contact avec l’association de patients ou le centre de traitement des troubles de la coagulation le plus proche pour poser des questions et discuter des options qui s’offrent à vous. Les associations de patients et les centres de traitement peuvent être localisés sur le site Internet de la FMH à l’adresse suivante : wfh.org/fr/soutien-sur-le-plan-local/.
Les personnes atteintes de troubles de la coagulation doivent être suivies dans un centre de traitement spécialisé dans le diagnostic et le traitement des troubles de la coagulation, où elles bénéficieront d’une prise en charge optimale et obtiendront les informations les plus pertinentes.
Une alimentation saine et une activité physique régulière permettent de conserver un corps sain et fort. L’exercice physique peut également contribuer à réduire le stress, l’anxiété et la dépression, ainsi que la fréquence et la gravité des saignements articulaires.
Les personnes atteintes d’un trouble de la coagulation sévère et qui sont vulnérables aux saignements articulaires devraient éviter les activités et les sports à fort impact tels que le football, la lutte et la planche à roulettes. Idéalement, un médecin ou un physiothérapeute compétent et expérimenté devrait prescrire les exercices aux personnes atteintes de troubles de la coagulation.
Une bonne hygiène bucco-dentaire est essentielle pour prévenir les caries et les maladies des gencives. Pour les personnes souffrant de troubles de la coagulation, il est très important de maintenir une bonne santé dentaire afin de réduire la nécessité d’une chirurgie dentaire, qui peut être compliquée par des saignements excessifs ou prolongés. Les personnes atteintes de troubles de la coagulation doivent :
Les procédures invasives, telles que le détartrage, les extractions ou les traitements du canal radiculaire, peuvent provoquer des saignements chez les personnes atteintes de troubles de la coagulation. Quelle que soit l’intervention, le dentiste doit consulter le centre de traitement des troubles de la coagulation afin de déterminer le risque possible encouru par la personne et d’élaborer un plan adéquat pour prévenir ou traiter les saignements. Des médicaments peuvent être nécessaires avant l’intervention pour prévenir les saignements et assurer une intervention et un rétablissement sans complications.
Les personnes atteintes de troubles de la coagulation doivent être vaccinées. Il est préférable d’administrer certains vaccins par voie sous-cutanée (sous la peau) plutôt que par voie intramusculaire afin d’éviter le risque de saignement associé. Il convient d’en discuter avec le médecin qui suit le patient pour le trouble de la coagulation concerné.
Il est important de vacciner contre l’hépatite A et B toute personne traitée avec du plasma frais congelé ou un produit n’ayant pas fait l’objet d’une inactivation virale. Les membres de la famille qui participent aux traitements doivent également être vaccinés, bien que cette précaution soit moins importante dans le cas des personnes qui utilisent des produits viro-inactivés.
Faites approuver tous vos médicaments par votre médecin. Il convient d’éviter certains médicaments en vente libre, car ils interfèrent avec la coagulation. Les personnes atteintes de troubles de la coagulation ne doivent pas prendre d’aspirine (acide acétylsalicylique) ou d’anti-inflammatoires non stéroïdiens (tels que l’ibuprofène et le naproxène) sans avis médical.
Les personnes atteintes de troubles de la coagulation doivent toujours avoir sur elles les informations sur leur maladie, le traitement nécessaire, ainsi que le nom et le numéro de téléphone de leur médecin ou de leur centre de traitement. En cas d’urgence, un bracelet d’alerte médicale ou tout autre moyen d’identification, comme la carte médicale internationale de la FMH, permet d’informer le personnel soignant de votre trouble de la coagulation. Avant de partir en voyage, trouvez l’adresse et le numéro de téléphone des centres de traitement des troubles de la coagulation à destination et conservez ces informations sur vous au cas où vous auriez besoin de soins. Les centres de traitement peuvent être localisés sur le site Internet de la FMH (wfh.org/fr/soutien-sur-le-plan-local/).
Les femmes atteintes d’un déficit en facteur de coagulation peuvent présenter davantage de symptômes que les hommes en raison du risque de saignement associé aux menstruations et à l’accouchement, tandis que les jeunes filles peuvent avoir des saignements abondants à la puberté.
Les femmes atteintes d’un déficit en facteur de coagulation peuvent avoir un flux menstruel plus abondant et/ou plus long, ce qui peut entraîner une carence en fer (faible taux de fer, ce qui entraîne faiblesse et fatigue) et/ou une anémie (faible taux de globules rouges).
Les femmes atteintes d’un déficit en facteur de coagulation doivent bénéficier d’un conseil génétique sur les risques de donner naissance à un enfant atteint dudit déficit, bien avant toute grossesse planifiée. Il est recommandé qu’elles consultent un obstétricien dès qu’elles pensent être enceintes. L’obstétricien doit travailler en étroite collaboration avec le personnel du centre de traitement des troubles de la coagulation afin de fournir les meilleurs soins possibles pendant la grossesse et l’accouchement, et de minimiser les complications potentielles pour la mère et le nouveau-né.
Les femmes atteintes d’un déficit en facteur de coagulation (tel que le déficit en facteur X, le déficit en facteur XIII et l’afibrinogénémie) peuvent être plus exposées au risque de fausse couche et de décollement placentaire (séparation prématurée du placenta de l’utérus qui perturbe la circulation du sang et de l’oxygène vers le fœtus). Ces femmes ont donc besoin d’un traitement tout au long de la grossesse pour éviter de telles complications.
Le principal risque lié à la grossesse est l’hémorragie post-partum. Les troubles de la coagulation sont associés à un risque accru de saignement, à la fois juste après l’accouchement et pendant plusieurs semaines suivant celui-ci. Les femmes atteintes d’un déficit en facteur de coagulation doivent donc collaborer avec leurs médecins (le spécialiste des troubles de la coagulation et l’obstétricien) pour élaborer un plan d’accouchement personnalisé. Ce plan doit couvrir toutes les étapes du travail, y compris la délivrance du placenta, afin de réduire le risque et la gravité des hémorragies. Le traitement varie selon chaque femme et dépend de ses antécédents hémorragiques personnels et familiaux, du diagnostic et de la gravité du trouble hémorragique, ainsi que du mode d’accouchement. Les femmes atteintes d’un déficit en facteur de coagulation doivent être invitées à contacter immédiatement leur professionnel de santé si les saignements post-partum sont trop abondants.
Dans certaines circonstances, les enfants nés de femmes atteintes d’un déficit en facteur de coagulation risquent également d’hériter de la maladie et de présenter des saignements. Il convient d’éviter les accouchements difficiles et prolongés, ainsi que les accouchements nécessitant l’utilisation d’instruments, tels que les forceps ou l’extraction par aspiration.



