








Français / Español / 简体中文 / русский / العربية / 日本語
© 2026 World Federation of Hemophilia
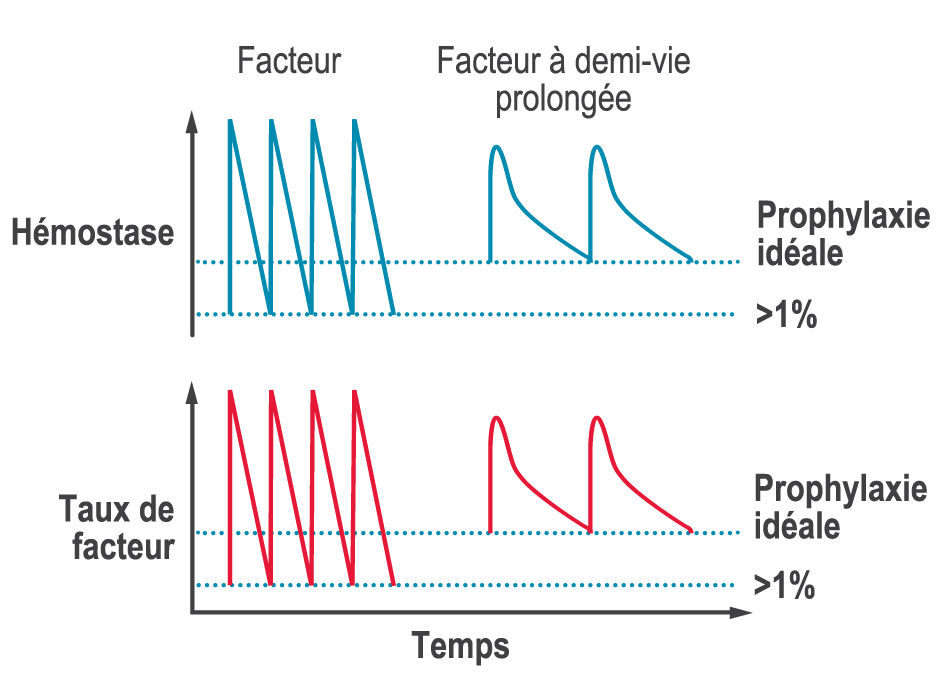
Pour l’hémophilie, le traitement recommandé est la prophylaxie à vie, traditionnellement fournie par des concentrés de facteur de coagulation (CFC) de remplacement, dès les 12 premiers mois de vie. L’accès à ces produits est limité dans de nombreux pays, et même dans les pays où l’accès aux concentrés de facteur n’est pas un problème, l’injection fréquente des concentrés constitue un fardeau important pour les personnes atteintes d’hémophilie, ainsi que pour les systèmes de santé.
L’une des principales limites des traitements avec facteur de remplacement est qu’après l’augmentation immédiate des taux de facteur après l’injection, ceux-ci diminuent fortement avant l’injection suivante (le fameux effet en dents de scie, voir schéma ci-dessous). Le manque de stabilité des taux de facteur entraîne une capacité moindre à former un caillot et, par conséquent, à prévenir et à stopper un saignement (hémostase), ce qui peut déboucher sur des saignements entre les différentes injections et des séquelles.
L’apparition éventuelle d’inhibiteurs constitue une complication grave due à une réponse immunitaire à l’injection de CFC. Ces inhibiteurs neutralisent l’efficacité du remplacement du facteur qui traite et prévient les saignements. Il existe bien quelques traitements en cas de présence d’inhibiteurs, mais la prise en charge est difficile et explique l’intérêt suscité par le développement de traitements sans facteur de remplacement afin de traiter les troubles de la coagulation.
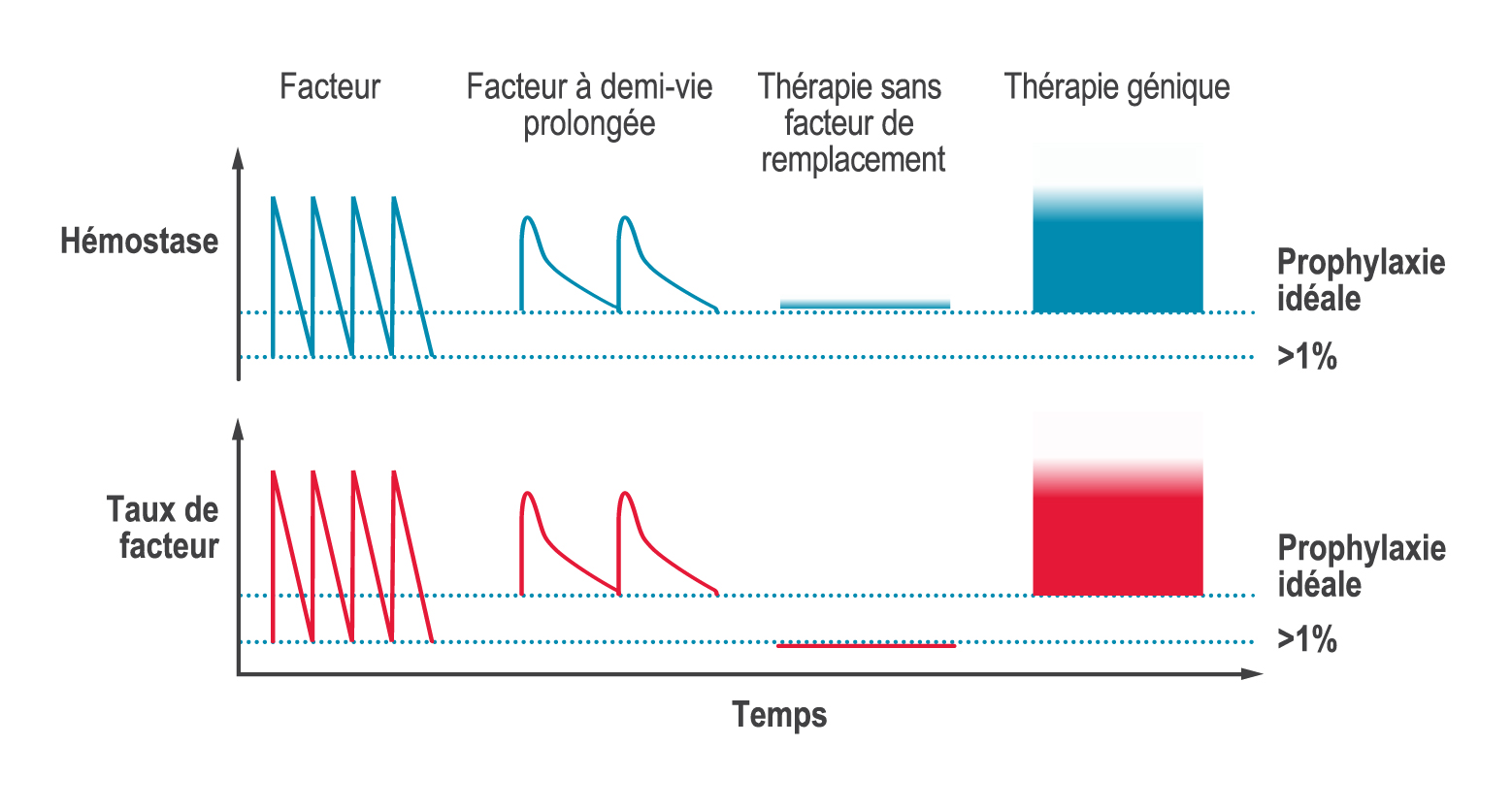
Les traitements sans facteur de remplacement constituent des approches thérapeutiques innovantes pour l’hémophilie, l’objectif étant de rééquilibrer l’hémostase sans avoir à remplacer le facteur de coagulation manquant. Ces traitements ciblent des étapes dans la cascade de coagulation, et l’approche est donc différente d’un simple remplacement du facteur VIII (hémophilie A) ou du facteur IX (hémophilie B) manquant. Plusieurs produits de sans facteur de remplacement ont été approuvés dans des pays du monde entier, notamment un anticorps bispécifique pouvant se substituer au facteur VIII dans la cascade de coagulation, et une thérapie de rééquilibrage hémostatique qui bloque un facteur d’anticoagulation d’origine naturelle. D’autres traitements sans facteur de remplacement sont en phase 3 d’essais cliniques.
L’hémostase est un savant équilibre entre un excès de coagulation (thrombose) et un manque de coagulation (saignement). Dans le cas de l’hémophilie, il manque un facteur de coagulation essentiel et la balance de l’hémostase penche du côté d’une tendance hémorragique plus forte. L’objectif des thérapies sans facteur de remplacement est de prévenir tout saignement en augmentant la capacité hémostatique (la faculté du sang à coaguler en cas de lésion vasculaire), plutôt qu’en augmentant le taux de facteur.
Les traitements par anticorps bispécifique sont des traitements sans facteur de remplacement et sont utilisés pour traiter l’hémophilie A. Les anticorps bispécifiques sont des protéines en forme de Y qui établissent un lien sélectif avec d’autres protéines. Ces anticorps bispécifiques peuvent se lier à deux protéines différentes en même temps.
Dans le cas de l’hémophilie A, l’anticorps agit comme un lien entre le facteur IXa et le facteur X, ce qui aide le sang à coaguler plus efficacement. Le lien mis en place permet d’imiter la fonction du facteur VIII activé manquant (le facteur VIIIa présente ce qu’on appelle une activité mimétique).
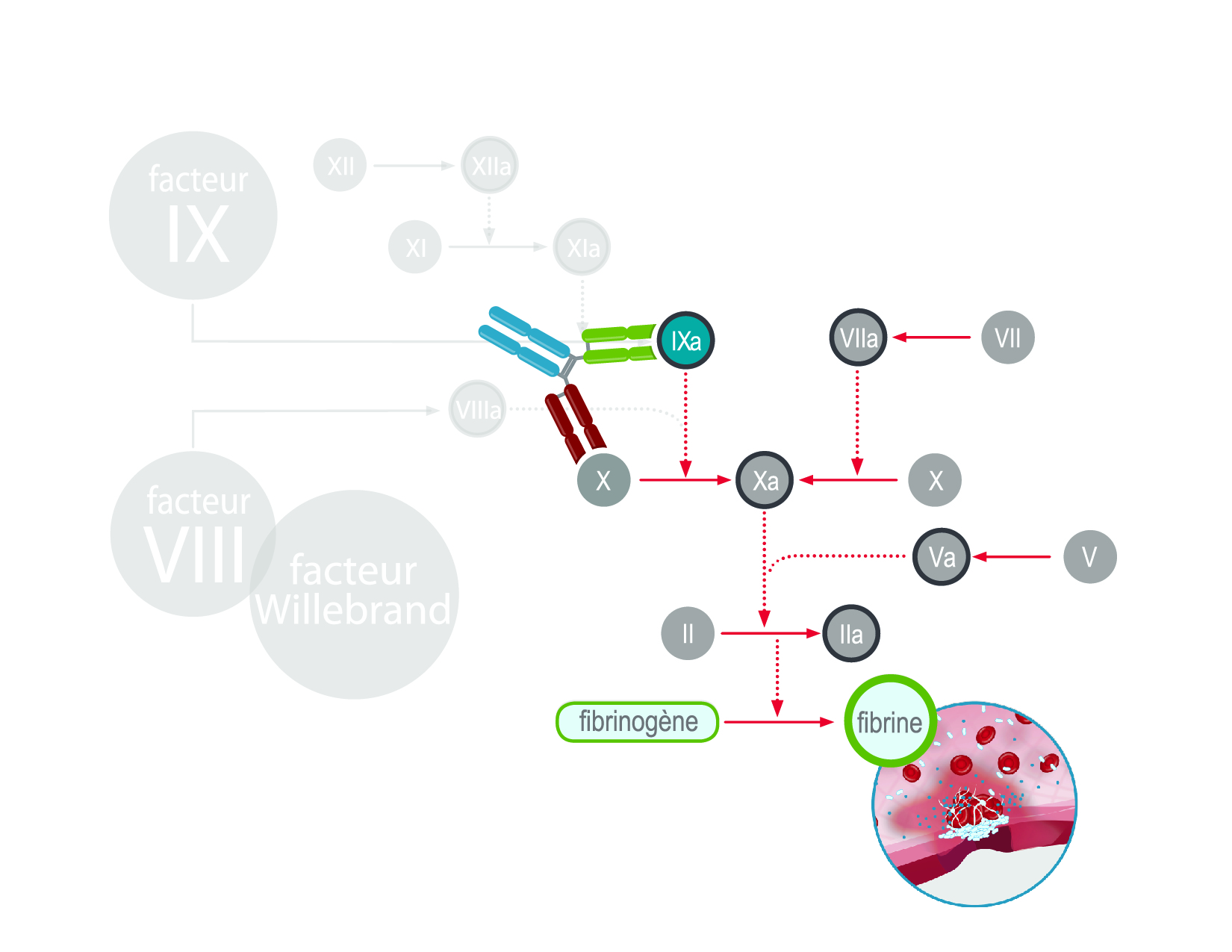
À partir de 2024, un traitement par anticorps bispécifique, l’emicizumab, imitant l’activité du facteur VIIIa, a été approuvé pour le traitement de l’hémophilie A. L’emicizumab est un anticorps monoclonal bispécifique, ce qui signifie qu’il a été produit en laboratoire et conçu pour reconnaître et se lier au facteur IX activé (FIXa) d’un côté au facteur X (FX) de l’autre côté. L’emicizumab permet ainsi d’assurer la fonction naturelle du facteur VIII activé (FVIIIa), qui correspond au facteur manquant ou déficient dans l’hémophilie A et qui est nécessaire pour permettre la coagulation sanguine. Dans la mesure où cette molécule ne remplace pas la fonction du FIX manquant, elle n’est pas indiquée pour l’hémophilie B. D’autres thérapies font l’objet d’essais cliniques.
Le recours à l’emicizumab est approuvé pour les personnes avec inhibiteurs contre le facteur VIII, et pour les personnes sans inhibiteurs atteintes d’hémophilie sévère (activité du FVIII < 1 %) ou modérée (activité du FVIII entre 1 et 5 %) avec un phénotype hémorragique sévère. L’emicizumab peut être utilisé dans tous les groupes d’âge (enfants et adultes).
Bien que l’emicizumab soit très efficace dans la prévention des saignements, il ne peut être utilisé pour traiter les hémorragies actives ou imprévues. Ce sont les professionnels de santé spécialisés dans le traitement de l’hémophilie qui doivent déterminer les dosages des produits, comme les agents de contournement et le facteur VIII, afin de traiter les différents types de saignements chez un patient sous emicizumab. Les personnes atteintes d’hémophilie sont invitées à observer le protocole d’urgence établi avec leur médecin hématologue pour traiter tout saignement imprévu. En effet, quelques personnes utilisant de fortes doses de concentrés de complexe prothrombinique activé (PCCA) associé à l’emicizumab ont eu des effets secondaires pouvant potentiellement engager le diagnostic vital, notamment des microangiopathies thrombotiques et des thromboembolies. Les spécialistes de l’hémophilie doivent éviter l’association de PPCA et d’emicizumab, sauf absence de toute autre alternative thérapeutique, comme le facteur VIIa.
L’emicizumab ne peut être évalué par certains tests de laboratoire utilisés habituellement pour mesurer la capacité de coagulation sanguine d’un individu, ce qui entraîne des résultats erronés. Avant de procéder à tout test de laboratoire mesurant la coagulation sanguine, les personnes atteintes d’hémophilie sous emicizumab doivent informer impérativement les professionnels de santé du traitement qu’ils suivent. Il est indispensable de le faire savoir pour éviter toute mauvaise interprétation des résultats des tests qui pourraient avoir une incidence sur la prise de décision thérapeutique.
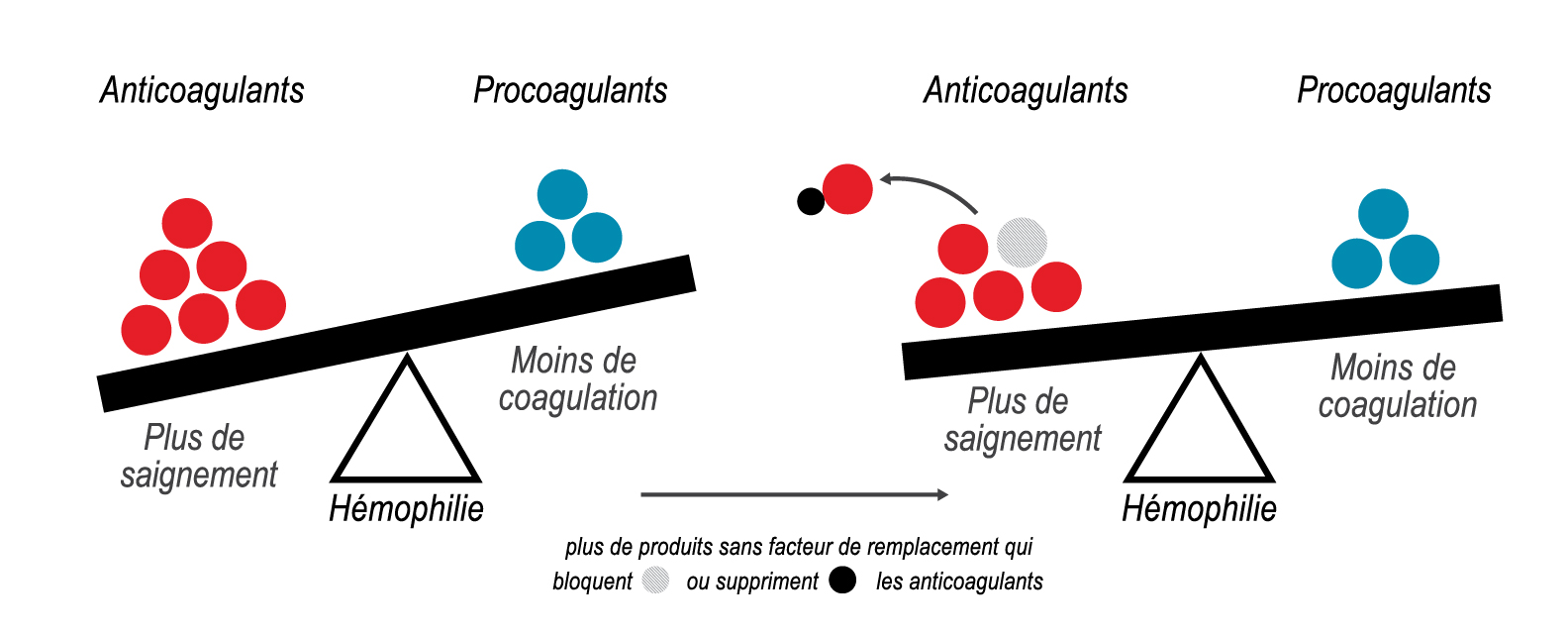
L’organisme dispose de plusieurs mécanismes pour éviter de prolonger plus que nécessaire le processus de coagulation ou de provoquer inutilement un tel processus, et pour, par conséquent, éviter une coagulation excessive. L’un des mécanismes consiste notamment à utiliser des anticoagulants naturels pour freiner l’action de la coagulation.
En cas de blessure, l’organisme arrête le saignement en activant des facteurs de coagulation déjà présents dans le sang et en générant de la thrombine. Les personnes atteintes d’hémophilie n’ont pas ou peu de facteur de coagulation VIII (hémophilie A) ou de facteur IX (hémophilie B), et la production de thrombine est faible, de sorte que le sang ne peut pas coaguler efficacement. En d’autres termes, chez les personnes atteintes d’hémophilie, il existe un déséquilibre entre les facteurs qui aident le sang à coaguler (facteurs de coagulation) et les facteurs qui empêchent la coagulation (facteurs d’anticoagulation). Les traitements par rééquilibrage de l’hémostase permettent de rétablir cet équilibre en diminuant les taux de facteurs anticoagulants, ce qui contribue à prévenir les hémorragies et à rétablir une coagulation sanguine normale.
En 2023, un traitement par rééquilibrage de l’hémostase, le concizumab, a été approuvé au Canada pour les personnes atteintes d’hémophilie B avec inhibiteurs. Le concizumab est un anticorps monoclonal qui ciblent un facteur naturel d’anticoagulation appelé inhibiteur de la voie du facteur tissulaire (TFPI). En inhibant le TFPI (ou anti-TFPI), ces médicaments augmentent la production de thrombine et la coagulation sanguine chez les personnes atteintes d’hémophilie A et B, avec ou sans inhibiteurs.
D’autres thérapies de rééquilibrage font l’objet d’essais cliniques de phase 3. L’un d’entre eux est un petit ARN interférent (siRNA) qui inhibe la production d’un autre coagulant, l’antithrombine. Cela augmente la production de thrombine et entraîne une augmentation de la coagulation sanguine chez les personnes atteintes d’hémophilie A et B, avec ou sans inhibiteurs.
L’objectif des produits sans facteur de remplacement est de rééquilibrer l’hémostase, aussi bien chez les personnes atteintes d’hémophilie A ou B, sans avoir à injecter de concentrés de facteur de coagulation (CFC). Bien que les thérapies sans facteur de remplacement permettent de réduire le degré de sévérité de l’hémophilie, ils ne guérissent pas et il est parfois nécessaire de recourir à un traitement à la demande en cas de saignement imprévu.
Les personnes atteintes d’hémophilie ayant recours à une thérapie sans facteur de remplacement sont invitées à :
Autres points importants :
