


Français / Español / 简体中文 / русский / العربية / 日本語
© 2026 World Federation of Hemophilia
В клинических исследованиях обычно участвуют добровольцы, участники или задействуют собранные у людей образцы (крови или других тканей) с целью расширения медицинских знаний. Существует два основных вида клинических исследований: клинические испытания (также называемые интервенционными исследованиями) и наблюдательные исследования.
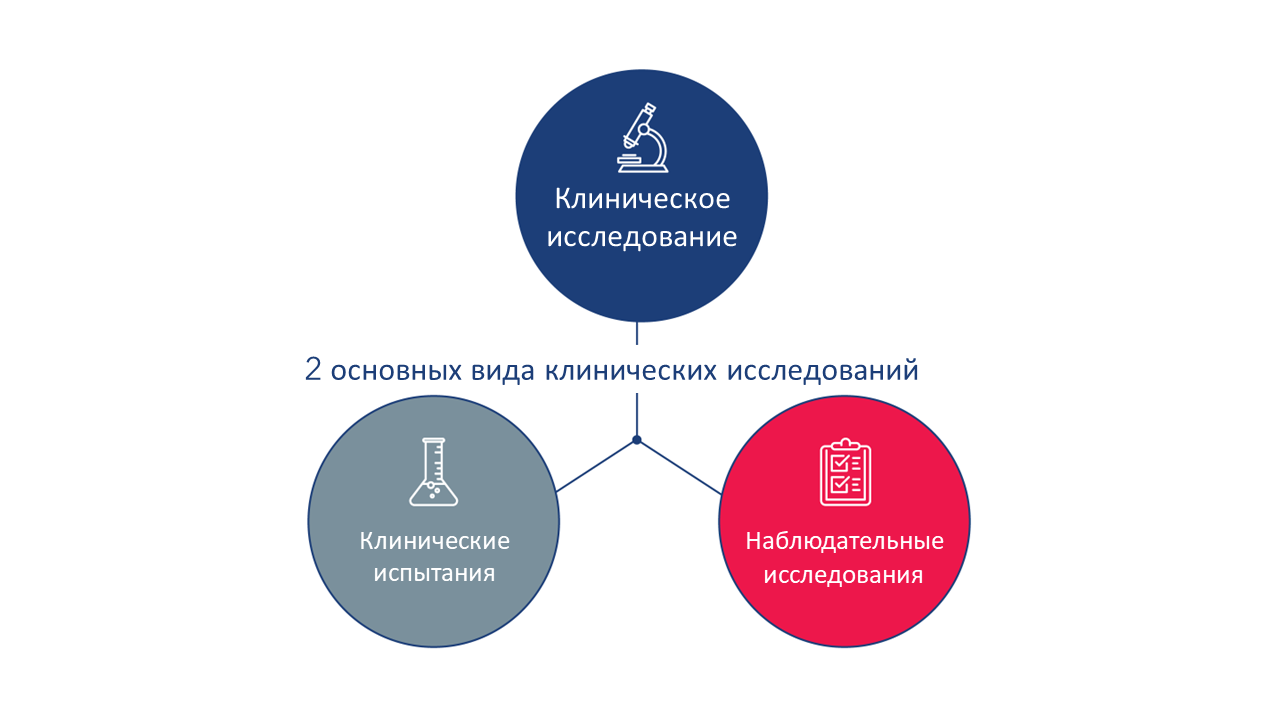
Клиническое испытание — это вид исследования, в ходе которого специалисты тестируют новые способы профилактики, выявления или лечения заболеваний. Участники клинического испытания подвергаются конкретным вмешательствам в соответствии с подробным протоколом соответствующего клинического испытания. Такими вмешательствами могут быть медицинские продукты, такие как лекарства или устройства, хирургические процедуры или изменения в поведении, например, диета участника.
В рамках клинических испытаний новый метод лечения может сравниваться с уже существующим стандартным методом, с плацебо или отсутствием вмешательства. Клинические испытания являются основным методом, который используют исследователи, чтобы выяснить, является ли новое лечение безопасным и эффективным. Чтобы лекарственное средство стало препаратом, который могут назначать врачи, оно должно пройти ряд клинических испытаний, известных как фаза 1, фаза 2 и фаза 3. После фазы 3 данные клинических испытаний представляются в регулирующий орган, который затем определяет, следует ли разрешить применение препарата.
Существуют различные виды клинических испытаний:
При описании клинических испытаний часто используются следующие термины:
Наблюдательное исследование — это вид исследования, в котором исследователи наблюдают за эффектами вмешательства в группе участников, не производя вмешательства самостоятельно. Исследователи не назначают участникам конкретные вмешательства, как в клинических испытаниях, но участники могут получать лечение, которое уже является частью их обычного медицинского обслуживания. Затем исследователи могут оценить связи между вмешательством и клиническими исходами среди людей, которые подвергаются вмешательству в рамках стандартного лечения. Такие результаты могут повлечь за собой дальнейшее исследование в рамках клинического испытания. Существует несколько различных видов наблюдательных исследований. Регистр пациентов — это один из видов наблюдательных исследований.
Регистр пациентов — это организованная система, в которой используются методы наблюдательного исследования для сбора данных о методах лечения, клинических результатах и благополучии населения, определяемом конкретным заболеванием, состоянием или экспозицией.
Для сообщества гемофилии важны два регистра пациентов:
1. Всемирный регистр коагулопатий
Всемирный реестр коагулопатий (ВРК (WBDR)) — это интернет-система ввода данных, которая обеспечивает платформу для сети центров лечения гемофилии по всему миру, позволяющую организовать сбор единообразных и стандартизированных данных о пациентах, а также направлять клиническую практику. С информированного согласия пациента в ВРК хранятся обезличенные данные о его заболевании, такие как тип и степень тяжести гемофилии, симптомы, лечение и клинические результаты.
Медицинские работники, участвующие в ВРК, могут использовать ВРК для отслеживания и мониторинга прогресса своего пациента и руководства его клиническим лечением. Эти обезличенные и конфиденциальные данные также могут быть использованы для того, чтобы помочь исследователям ответить на важные вопросы о неравенстве в уровне медицинского обслуживания во всем мире и содействовать продвижению инициатив в области адвокации и политики здравоохранения.
Нажмите здесь для просмотра страницы ВРК.
2. Регистр генной терапии Всемирной федерации гемофилии
Благодаря международному сотрудничеству ВФГ разработала всемирный регистр пациентов, получающих генную терапию, — регистр генной терапии ВФГ (РГТ(GTR)). Цель РГТ — создать надёжную, соответствующую научным критериям базу данных, доступную для всех медицинских учреждений, занимающихся лечением людей с гемофилией, которые получают генную терапию, в любой точке мира. Данные, собранные с помощью РГТ ВФГ, будут использоваться для оценки долгосрочных результатов по безопасности и эффективности генной терапии у лиц с гемофилией.
Нажмите здесь для получения информации об РГТ ВФГ.
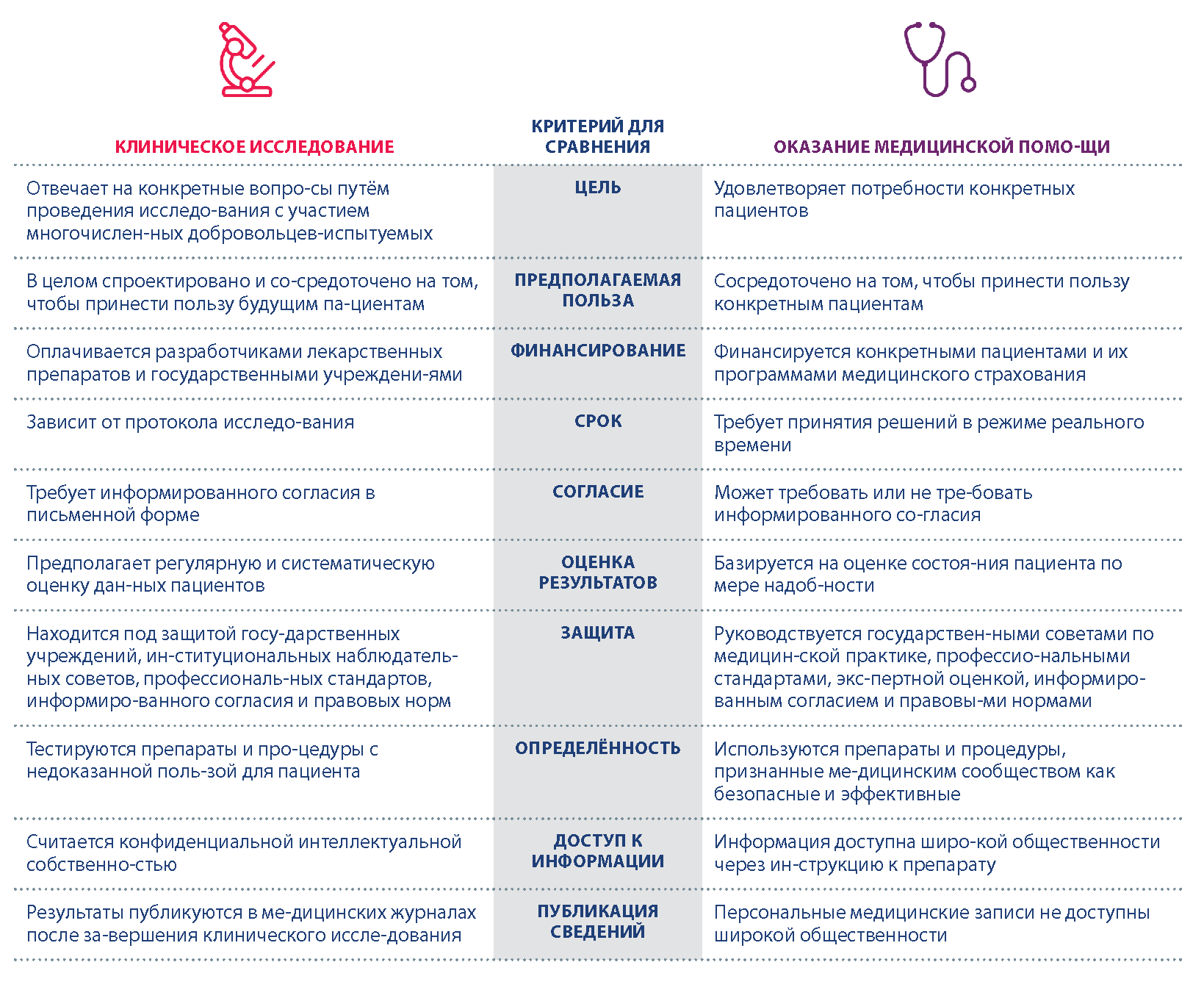
Следующая таблица может быть полезна для понимания того, чем клинические исследования отличаются от оказания медицинской помощи.
Для того, чтобы лекарственный препарат стал средством, которое могут назначать врачи, он должен сначала пройти через ряд фаз клинических испытаний, а затем получить оценку и одобрение регулирующего органа. Каждая фаза клинических испытаний имеет свою цель, и фазы проходят в порядке от фазы 1 до фазы 4.
Фаза 1.
Это — первый раз, когда новое исследуемое лекарственное средство испытывается на людях. Цель клинических испытаний фазы 1 — оценить безопасность и побочные эффекты, связанные с лечением. Они часто проводятся на небольшой группе здоровых добровольцев. Однако при редких заболеваниях, таких как гемофилия, испытания фазы 1 обычно проводятся на людях с гемофилией. Клинические испытания фазы 1 обычно длятся несколько месяцев, и примерно 70% лекарственных средств, протестированных в рамках фазы 1, переходят в следующую фазу. Клиническая разработка не выходит за рамки фазы 1 для остальных 30% препаратов, которые не показали свою безопасность в фазе 1.
В клинических испытаниях 1-й фазы генной терапии гемофилии принимают участие от 10 до 30 человек с гемофилией.
Фаза 2.
Цель клинических испытаний фазы 2 — продолжить проверку безопасности и побочных эффектов, связанных с лечением, а также протестировать эффективность и определить наиболее эффективные дозировки. Испытания фазы 2 обычно проводятся на большем количестве участников, чем испытания фазы 1, и включают людей, страдающих заболеванием. В некоторых ситуациях испытания фазы 1 и фазы 2 объединяются и называются фазой 1/2. Клинические испытания фазы 2 обычно длятся от нескольких месяцев до 2 лет. По оценкам, 30% лекарственных средств, прошедших испытания в рамках фазы 2, переходят в следующую фазу. Клинические разработки не выходят за рамки фазы 2, если не доказана эффективность лечения или имеются проблемы с безопасностью.
В клинических испытаниях 2-й фазы генной терапии гемофилии участвуют от 10 до 50 человек с гемофилией. Это число меньше, чем при других заболеваниях, поскольку гемофилия – редкое заболевание. Клинические испытания I и 2 фазы часто объединяются для редких заболеваний, таких как гемофилия. Объединяя фазы испытаний, производители лекарственных средств надеются ускорить сроки разработки новых препаратов.
Фаза 3.
Цель клинических испытаний фазы 3 — подтвердить эффективность лечения, провести мониторинг побочных эффектов и сравнить новый препарат со стандартными или аналогичными средствами. Испытания фазы 3, иногда называемые «опорными испытаниями», проводятся на большом количестве людей с заболеванием и в разных местах (в масштабе страны или на международном уровне, в зависимости от исследования). Клинические испытания фазы 3 иногда рандомизированы и зачастую проводятся в двойном слепом режиме. Клинические испытания фазы 3 обычно длятся от 1 до 4 лет и являются последним этапом перед подачей заявки в регулирующий орган для получения разрешения. Примерно 25% исследований фазы 3 переходят в фазу 4.
Данные опорного исследования фазы 3, а иногда и данные клинических исследований фаз 1 и 2 представляются на рассмотрение в регулирующий орган. Он, в свою очередь, проводит независимый анализ безопасности и эффективности терапии и принимает решение о том, разрешена ли терапия для использования пациентами или нет.
Клинические испытания 3-й фазы генной терапии гемофилии охватывают 50-150 человек с гемофилией и проводятся в исследовательских центрах по всему миру.
Фаза 4.
Фаза 4, иногда называемая постмаркетинговыми исследованиями, проводится после того, как новая терапия получила одобрение регулирующих органов и стала доступно для пациентов. Эти исследования позволяют ученым собрать дополнительную информацию о долгосрочных рисках (включая редкие побочные эффекты) и преимуществах терапии, а также об оптимальном применении в реальных условиях.
Дополнительную информацию см. в PDF-документе «Как новые методы лечения тестируются в ходе клинических испытаний».

Каждое клиническое испытание имеет подробный, всеобъемлющий план проведения исследования, называемый протоколом. Протокол клинического испытания разрабатывается для того, чтобы ответить на конкретные вопросы исследования и защитить здоровье участников исследования. Перед началом клинического испытания протокол рассматривается и утверждается регулирующим органом. Информация, содержащаяся в протоколе клинического испытания, включает в себя нижеследующее:
Программа клинической разработки нового метода терапии или вмешательства определяет, является ли новая терапия эффективной и безопасной. В протоколе клинического испытания первичный конечный критерий — это запланированный показатель результата, который является наиболее важным для оценки вмешательства или способа лечения. В зависимости от фазы клинического испытания первичный конечный критерий может быть направлен на безопасность, например, связанные с лечением нежелательные явления и/или изменения по сравнению с исходным уровнем в клинических лабораторных тестах; или же критерий может быть сфокусирован на эффективности, например, годовой частоте кровотечений или уровне фактора.
Примеры конечных критериев безопасности и эффективности в клинических испытаниях при гемофилии (включая испытания генной терапии):
| Конечные критерии безопасности | Конечные критерии эффективности |
|---|---|
|
|
Очень важно понимать потенциальные преимущества и риски участия в клиническом испытании.
| Потенциальные преимущества | Потенциальные риски |
|---|---|
| Вы можете получить доступ к новому методу лечения до его появления и стать одним из первых, кто воспользуется его преимуществами | Вы можете столкнуться с нежелательными побочными эффектами нового лечения |
| Вы будете пользоваться поддержкой команды специалистов по лечению гемофилии, которые будут внимательно следить за вашим здоровьем | Новый метод терапии может не сработать для вас, или же вы можете получить плацебо, если это плацебо-контролируемое исследование |
| У вас будет возможность играть активную роль в сохранении вашего здоровья и улучшить собственную стратегию лечения гемофилии. | Изучаемый новый метод терапии может оказаться не лучше, чем ваш уже имеющийся стандарт лечения. |
| Вы можете помочь будущим пациентам с диагнозом гемофилия, внеся свой вклад в разработку потенциального метода терапии. | Испытание может отнять у вас больше времени, чем ваше обычное лечение; возможно, вам придется чаще посещать врачей и сдавать анализы |
Потенциальным участникам клинического испытания необходимо помнить, что его целью является изучение нового метода лечения или вмешательства.
Пациенты приглашаются к участию в клинических испытаниях через свою команду по оказанию медицинской помощи, исходя из критериев соответствия требованиям данного исследования. Под критериями соответствия требованиям понимаются основные требования, которые должны быть выполнены для участия в клиническом испытании. Эти критерии помогают обеспечить безопасность участников и гарантировать, что на конкретные исследовательские вопросы, прорабатываемые в рамках клинического испытания, можно будет получить точные ответы.
Каждое исследование имеет как критерии включения, так и критерии исключения.
Для клинических испытаний гемофилии общими факторами, определяющими соответствие требованиям, являются степень тяжести и тип гемофилии, возраст, ингибиторный статус, а также история профилактики. Каждое клиническое испытание уникально, и критерии соответствия требованиям варьируются от исследования к исследованию. Многие клинические испытания начинаются сначала на взрослых, перед тем, как изучать новый метод лечения на детях. По этой причине очень часто возраст указывается в качестве критерия включения.
Хотите узнать больше о критериях включения?
Люди с гемофилией, заинтересованные в участии в клиническом исследовании, проходят «процесс отбора», в ходе которого исследовательская группа определяет, соответствует ли данный человек критериям отбора для участия в исследовании. Процесс отбора будет включать изучение истории болезни и текущего медицинского состояния ЛСГ, а также обсуждение ролей и обязанностей участников, потенциальных рисков и преимуществ участия.
Важно понимать, что не у всех, кто заинтересован в клиническом исследовании, будет возможность принять в нём участие. Это может произойти потому, что какой-то аспект истории болезни человека не соответствует критериям включения в исследование (например, в исследование могут быть включены только ЛСГ определённого возраста). Кроме того, в каждом клиническом исследовании будет указано предполагаемое количество участников для запланированного набора; когда клиническое исследование наберёт необходимое количество людей, оно прекратит приём дополнительных участников исследования.
Рассмотрим пример (вымышленный) критериев приемлемости для клинических испытаний по гемофилии: Пример 1 Генная терапия при гемофилии
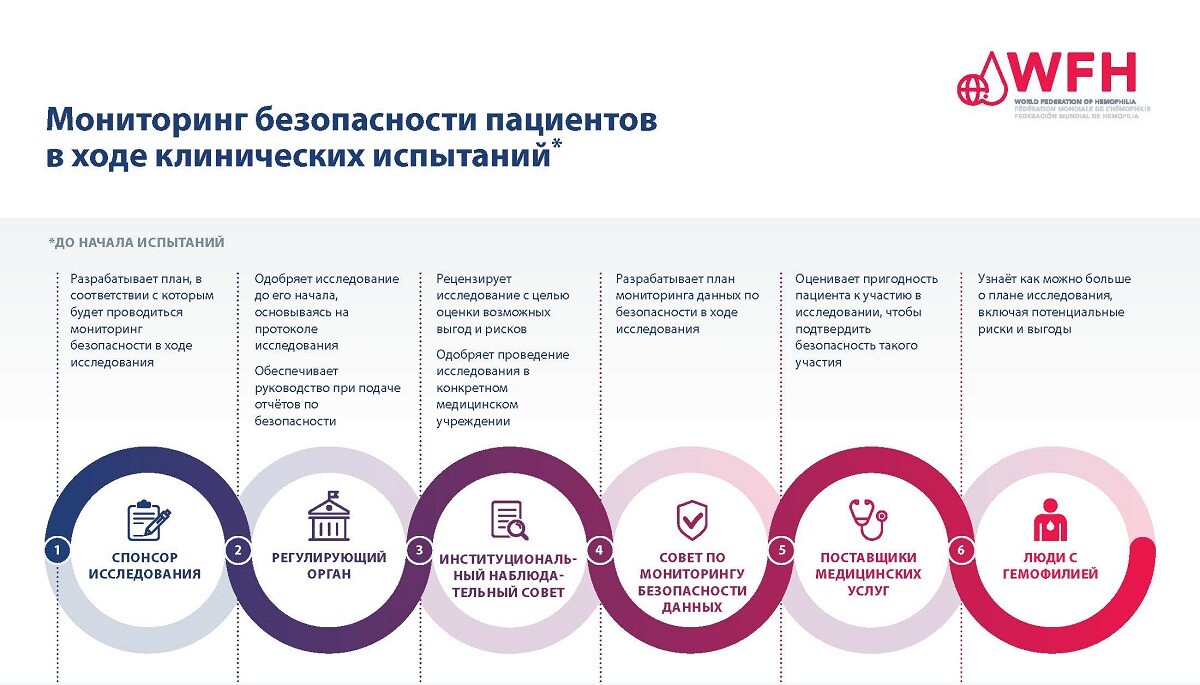
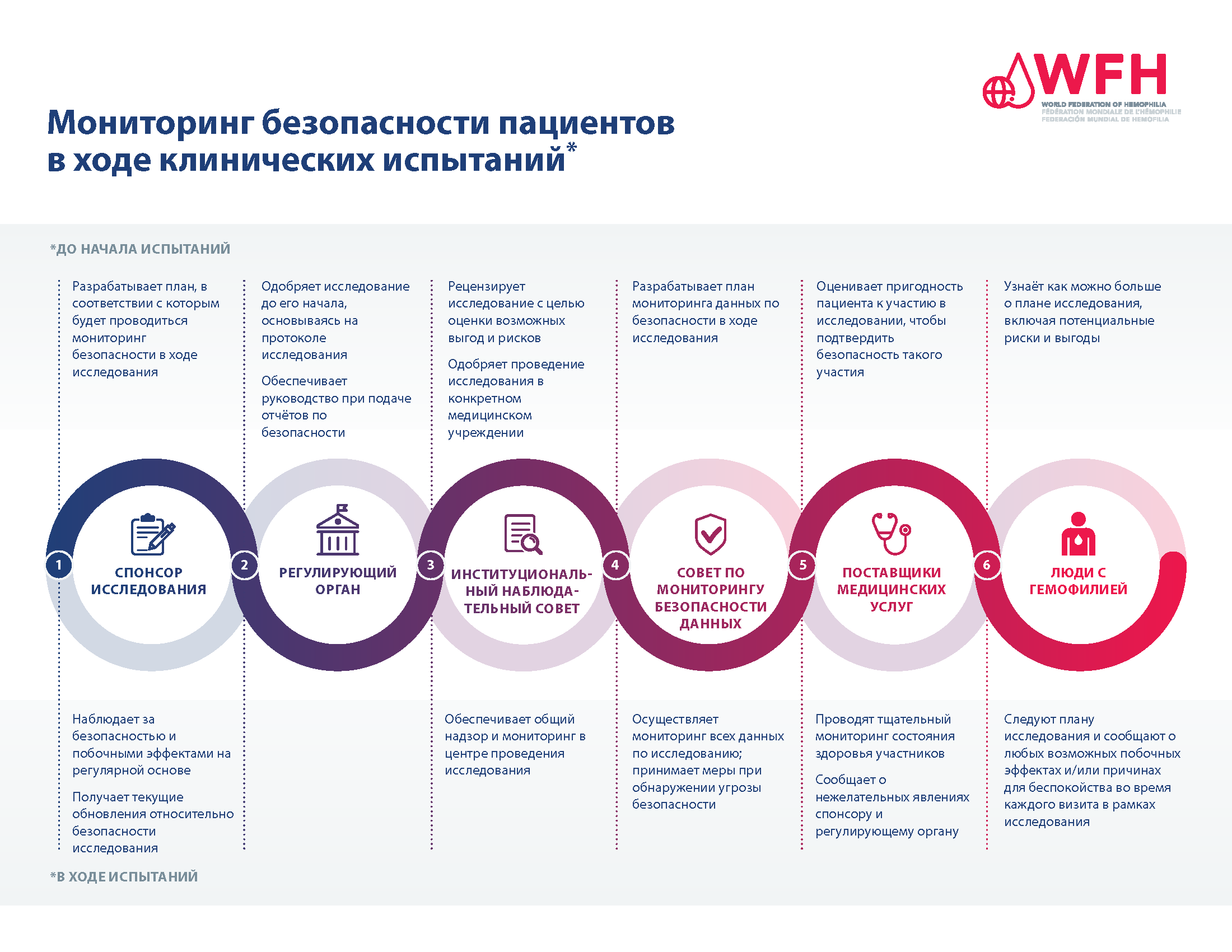
Обеспечение безопасности пациентов имеет первостепенное значение в процессе клинических исследований. Для обеспечения безопасности участников клинических испытаний существует множество уровней утверждения и мониторинга исследований, а также система сбора и предоставления отчётности о результатах в области безопасности по ходу исследования.
Независимо от этапа клинического испытания каждое исследование должно быть рассмотрено и одобрено регулирующим органом, прежде чем в него будут вовлечены пациенты. Спонсор исследования, часто производитель исследуемого метода лечения, разрабатывает протокол исследования, который включает раздел о том, как в ходе исследования будет контролироваться безопасность терапии. Затем протокол исследования должен быть утверждён регулирующим органом, таким как Управление по контролю за продуктами и лекарствами США (FDA) или Европейское агентство по лекарственным средствам (EMA). После утверждения исследования и определения мест проведения исследования участвующие в нем учреждения должны запросить и получить одобрение протокола исследования в Экспертном совете больницы или центра лечения гемофилии, где оно будет проводиться.
После утверждения исследования безопасность участвующих в нем пациентов контролируется на протяжении всего исследования Независимым комитетом по мониторингу данных, спонсором или компанией-производителем исследуемого метода лечения, а также участвующими в исследовании медицинскими работниками. ЛСГ, участвующие в исследовании, также играют определенную роль. ЛСГ, участвующие в исследованиях, должны информировать исследовательскую группу в своём лечебном центре, если у них возникают какие-либо побочные эффекты или события, связанные с безопасностью.
Дополнительную информацию см. в PDF-документе «Мониторинг безопасности пациентов в ходе клинических исспытаний».
Каждый участник, желающий принять участие в клиническом испытании, должен до включения в исследование подписать форму информированного согласия (либо родитель может подписать форму согласия за своего ребёнка в случае участия несовершеннолетних). Информированное согласие предоставляет потенциальным участникам информацию об испытании и объясняет потенциальные риски и преимущества, связанные с участием в исследовании, до того, как человек примет решение об участии. Процесс получения информированного согласия включает в себя обсуждение с исследовательской группой до подписания формы.
В целом процесс получения информированного согласия включает в себя нижеследующее:
Ниже приводится подробный список, составленный регуляторным органом, в данном примере — FDA (Управлением по контролю за продуктами и лекарствами США). Как часть процесса получения информированного согласия, эта информация должна быть предоставлена каждому потенциальному субъекту исследования перед его включением в клиническое испытание:
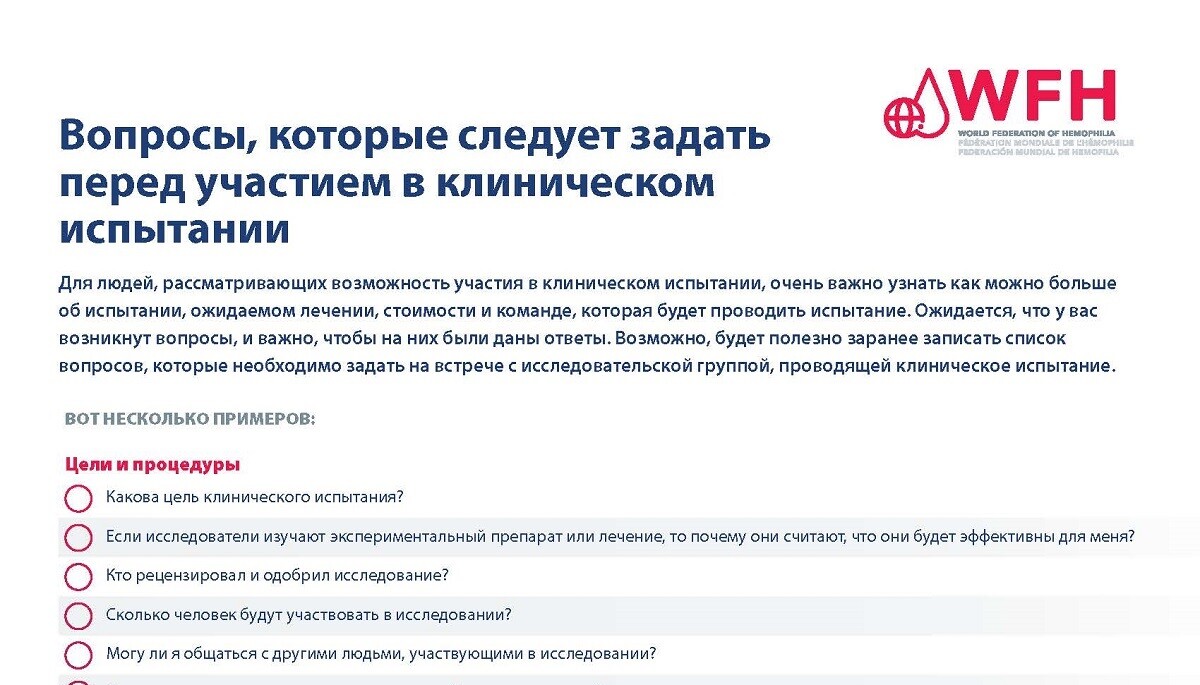
Для людей, рассматривающих возможность участия в клиническом испытании, чрезвычайно важно как можно больше узнать об испытании, о рисках и преимуществах для участников, об обязанностях участников, ожидаемом медицинском обслуживании и команде, которая будет проводить испытание. Ожидается, что у вас возникнут вопросы, и принципиально важно, чтобы на них были даны ответы. Возможно, перед встречей с исследовательской группой, проводящей клиническое испытание, будет полезно составить список вопросов, которые следует задать.
Для дополнительной информации см. PDF-документ «Вопросы, которые следует задать перед участием в клиническом испытании».

Члены медицинской бригады играют важную роль в обеспечении того, чтобы потенциальные участники исследования полностью понимали, что будет означать участие в клиническом испытании.
| Компоненты процесса получения информированного согласия | Удовлетворение потребностей потенциальных участников клинических испытаний |
|---|---|
|
|
|
|
|
|
|
|
|
|
У людей, рассматривающих возможность участия в клиническом испытании, могут возникнуть вопросы о медицинской информации и конфиденциальности. Конфиденциальность означает сохранение в тайне индивидуально идентифицируемой информации о состоянии здоровья лиц, участвующих в клинических испытаниях. Она включает в себя записи исследований, относящиеся к идентификации (например, имя), диагнозы, прогнозы, сведения о лечении или любую другую информацию, которую можно связать с участником. В случаях, когда результаты клинического испытания публикуются в рецензируемом журнале, информация, относящаяся к пациенту, обезличивается, и в таких отчётах персональные данные сохраняются в тайне.
В процесс клинических испытаний вовлекаются многие различные группы людей, которые имеют различные обязанности и роли. Участники и поставщики медицинских услуг играют важную роль на протяжении всего процесса клинического испытания. Важно, чтобы как участники, так и поставщики медицинских услуг полностью понимали свою роль.
Ниже приведены роли и обязанности каждой из этих групп.
Для получения дополнительной информации см. PDF-документ «Роли пациента и поставщика медицинских услуг в ходе клинических испытаний».

В каждой стране есть свой регулирующий орган со своими правилами или законами относительно проведения клинических испытаний. Регулирующий орган рассматривает и утверждает протоколы клинических испытаний до начала исследований и следит за тем, чтобы все клинические испытания соответствовали государственным законодательным нормам. Регулирующий орган взаимодействует с исследователями на протяжении всего процесса клинических испытаний и в конечном итоге анализирует все данные о безопасности и эффективности программы клинических разработок, чтобы определить, следует ли утвердить новый препарат/вмешательство и сделать его общедоступным.
Частное лицо, компания, учреждение, группа или организация, которая берёт на себя ответственность за инициирование, управление и/или финансирование клинического испытания.
Группа учёных, врачей, сотрудников, не связанных с наукой, а также специалистов по адвокации, которая рассматривает и утверждает подробный план клинического испытания. ЭСО призван защищать людей, которые принимают участие в клиническом испытании. В странах помимо США эта группа называется Этическим комитетом.
Группа независимых учёных, которые следят за безопасностью и достоверностью клинического исследования.
Лицо (лица), ответственное (ые) за клиническое испытание. Руководитель испытания, или «РИ», часто является врачом.
Лицо, ответственное за проведение клинических испытаний в соответствии с принципами надлежащей лабораторной практики (НЛП) и работающее под управлением руководителя клинического испытания.
Члены медицинской бригады, участвующие в проведении клинического испытания. В неё входят врачи, медсёстры, фельдшеры, фармацевты, учёные и другие лица, которые оказывают поддержку участникам на протяжении всего процесса клинического испытания, проводят тесты, оценку/анализы во время посещений, собирают данные и выполняют все части протокола клинического испытания.
Лица, отвечающие критериям соответствия требованиям и участвующие в клиническом испытании.