The standard of care for hemophilia is life-long prophylaxis, traditionally provided through replacement clotting factor concentrates (CFCs), starting as early as the first year of life. Access to these products is limited in many countries, and even in countries where access to factor concentrates is not an issue, the frequent infusions are a significant burden on people with hemophilia and on healthcare systems.
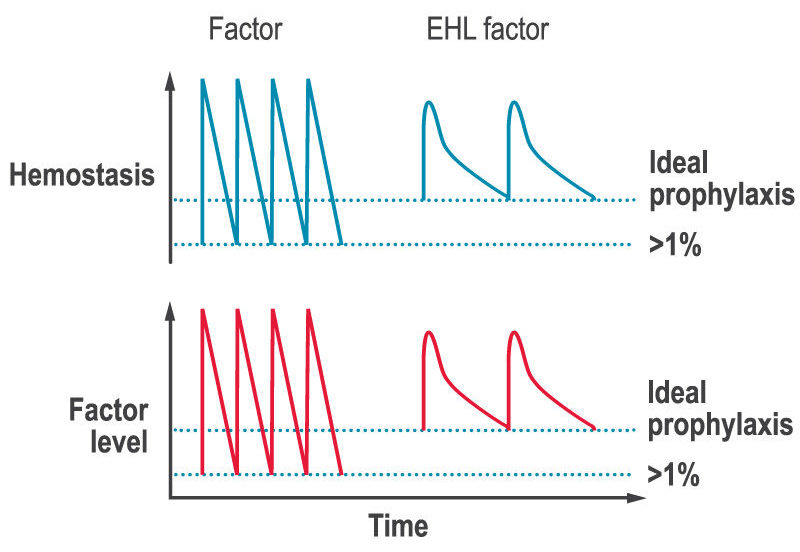
A key limitation of factor replacement products is, after the immediate rise in factor levels after an infusion, they then drop to low levels before the next infusion (the saw-tooth response, see figure below). The lack of stable factor levels results in a decrease in ability to form clots and therefore to prevent and stop bleeds (hemostasis), which can lead to bleeds and joint damage between infusions.
Inhibitors are a serious potential complication of hemophilia caused by an immune response to CFCs that inhibit the effectiveness of replacement factor to treat and prevent bleeds. While some treatment options do exist for inhibitors, the challenges of managing hemophilia in the presence of inhibitors also contributed to the interest in developing novel therapies to treat bleeding disorders.
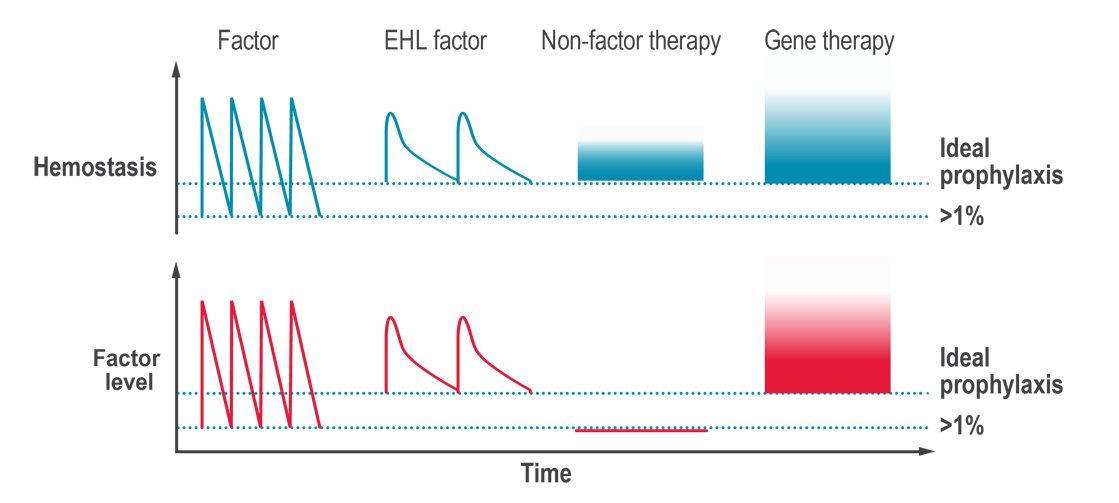
Non-factor replacement products are innovative treatment options for hemophilia that aim to rebalance hemostasis without the need for replacing the clotting factor that is missing. They target different points in the coagulation cascade, other than simply replacing the missing factor VIII (hemophilia A) or factor IX (hemophilia B). Multiple non-factor replacement products have been approved in countries around the world, including a bispecific antibody that can replace factor VIII in the clotting cascade, and a hemostatic rebalancing therapy that blocks a naturally occurring anticoagulation factor. Other non-factor replacement therapies are in phase 3 clinical trials.
Hemostasis is a fine balance between too much clotting (thrombosis) and not enough clotting (bleeding). In hemophilia, an essential clotting factor is lacking, and the hemostatic balance is tipped toward too much bleeding. The goal of non-factor replacement therapies is to prevent bleeds by raising hemostatic potential (how likely the blood is to clot if a blood vessel is injured), rather than by raising factor levels.
Bispecific antibody therapies are non-factor replacement therapies and are used to treat hemophilia A. Antibodies are Y-shaped proteins that can selectively bind to other proteins. Bispecific antibodies can bind to two different proteins at the same time.
In hemophilia A, the antibody acts as a bridge between two important proteins in the body during clotting (coagulation cascade), factor IXa (9a) and factor X (10), which helps the blood clot more efficiently: this mimics the function of the missing factor VIII (8), and therefore, these therapies are also referred to as factor VIII(a)-mimetics.
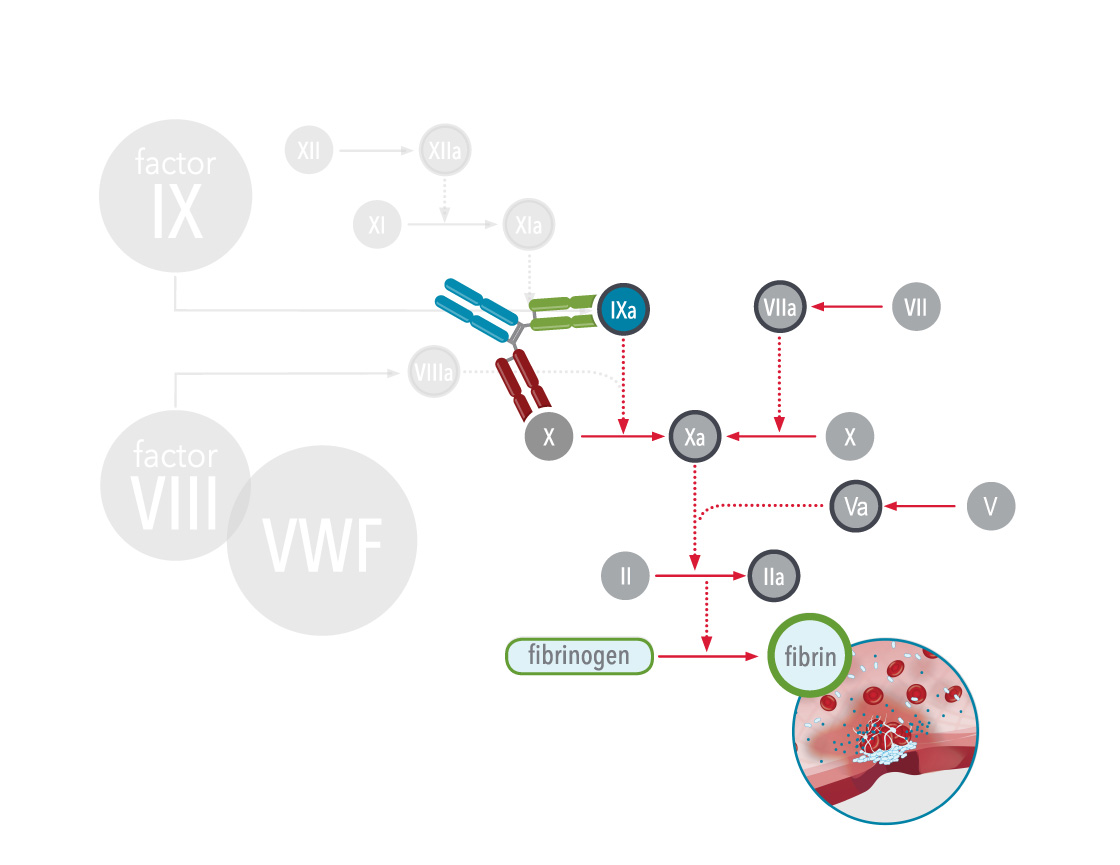
As of 2024, one bispecific antibody therapy, emicizumab, has been approved for treatment of hemophilia A. Emicizumab is a bispecific monoclonal antibody, which means it has been manufactured in a laboratory and designed to recognise and bind to activated factor IX (FIXa) on one arm and to factor X on the other arm. By doing so it carries out the natural function of activated factor VIII (missing or defective in hemophilia A) that is needed for effective clotting. Since it does not replace the function of missing factor IX it cannot be used to treat hemophilia B. Other therapies are in clinical trials.
Emicizumab is approved for use in people with hemophilia A with and without inhibitors who have severe disease (FVIII activity <1%) or moderate disease (FVIII activity between 1 and 5%) with severe bleeding phenotype. Emicizumab can be used in all age groups (newborn and older).
Although emicizumab is highly effective at preventing bleeds, it cannot be used to treat breakthrough, active or ongoing bleeds. Healthcare professionals experienced in the treatment of hemophilia must determine the appropriate dosing of agents, such as bypassing agents and factor VIII, to treat different kinds of bleeds in someone who is taking emicizumab. People with hemophilia should carefully follow the urgent action plan they establish with their hemophilia specialist to treat breakthrough bleeds. A few people using specific combinations of high doses of activated prothrombin complex concentrates (aPCCs) and emicizumab have experienced serious and potentially life-threatening side effects including thrombotic microangiopathy and thromboembolism. Hemophilia specialists should avoid using aPCCs and emicizumab at the same time unless no other treatment options, such as factor VIIa, are available.
Emicizumab interferes with certain laboratory tests that measure how well a person’s blood clots, leading to false readings. Before undergoing laboratory tests that measure blood clotting, people with hemophilia should inform any healthcare professionals that they are taking emicizumab. It is important for them to know so that they do not misinterpret the results of the tests, which could affect management decisions.
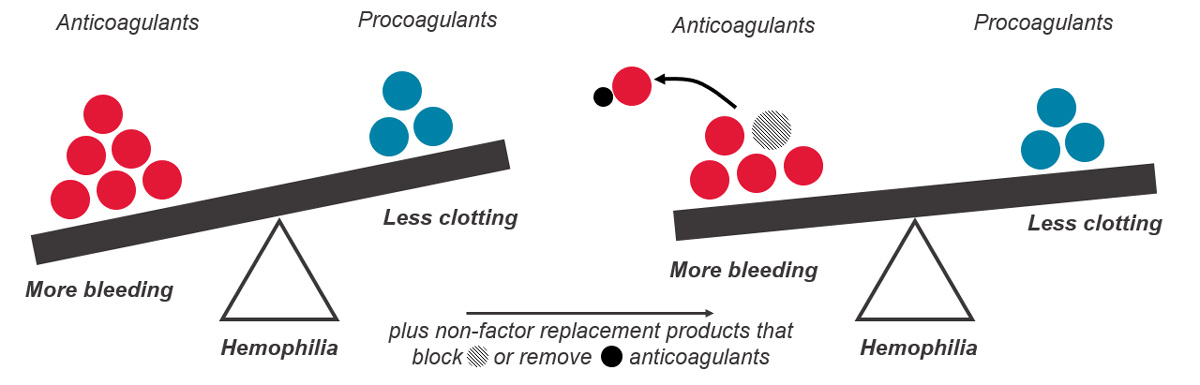
The body has several processes in place to prevent coagulation running longer than necessary or starting when it should not, to avoid too much blood clotting. One of these mechanisms includes natural anticoagulants that act as brakes to limit clotting.
When we are injured, our body’s natural system stops the bleeding through activation of the clotting factors already present in the blood and through thrombin generation. People with hemophilia have no or low levels of clotting factor VIII (hemophilia A), or factor IX (hemophilia B) and low thrombin generation, so their blood cannot clot effectively. In other words, in people with hemophilia, there is an imbalance between the factors that help the blood clot (clotting factors) and the factors that prevent clotting (anticoagulation factors). Rebalancing therapies help to restore this balance by decreasing the anticoagulation factor levels, which helps prevent bleeding events and restore normal blood clotting.
As of 2023, one hemostatic rebalancing therapy, concizumab, has been approved for use in Canada for people with hemophilia B with inhibitors. Concizumab is a monoclonal antibody that targets a natural anticoagulation factor called tissue factor pathway inhibitor (TFPI). By inhibiting TFPI (also known as anti-TFPI) these medications increase thrombin generation and blood clotting in people with hemophilia A and B, with and without inhibitors.
Other rebalancing therapies are in phase 3 clinical trials. One of these is a small interfering RNA (siRNA) that inhibits the production of another coagulant, antithrombin. This increases thrombin generation and results in increased blood clotting in people with hemophilia A and B, with and without inhibitors.
The potential of non-factor replacement products is to rebalance hemostasis, in people with hemophilia A or B, without the need for infusions of clotting factor concentrates (CFCs). While non-factor replacement products reduce the severity of hemophilia, these products do not provide a cure and occasional on-demand treatment for breakthrough bleeds may be required.
People with hemophilia who are taking a non-factor replacement product should:
Other important points:




